Chemical Kinetics
A knowledge of the kinetics of the reaction employed in an industrial process helps to maintain optimum conditions for the reaction so that the process becomes economically viable. Generally, chemical reactions that are fast with a high yield of products at reasonable costs are used in industrial processes.
The study of rates of reactions helps in understanding the mechanisms of reactions. An understanding of the mechanism of a biochemical reaction helps in analyzing how an enzyme works.
Reaction Rate
The rate of a reaction may be expressed in terms of the amount of reactants consumed or the amount of products formed in n given time. In other words, it is the change in concentration of the reactant or product with respect to the change in time after the reaction has been initiated.
Chemical kinetics class 12 chemistry notes
Since the rate of a reaction is influenced by temperature, the temperature of the reaction mixture should be held constant during the course of the reaction. If it is not possible to measure the concentration directly, then a change that is directly proportional to the concentration can be measured.
For example, the change in the number of moles per cubic meter or a change in the pressure of a gas, if a gas is involved in the reaction, can be measured.
Thus, the rate of a reaction can be defined as
⇒ \(\frac{change in the concentration of the product}{time taken}\)
or, \(\frac{change in the concentration of the reactant}{time taken}\)
Units of rate of reaction If the concentration is expressed in mol L-1 then the unit of the rate of a reaction is mol Vs. In a gaseous reaction, when the concentration of gases is expressed in terms of their partial p then the unit of the rate is atm s-1 or bar s-1.
Let us consider the thermal decomposition of nitrogen pentoxide N2O5, which gives the brown gas nitrogen dioxide and molecular oxygen.
\(\underset{\text { colourless }}{2 \mathrm{~N}_2 \mathrm{O}_5(\mathrm{~g})} → \underset{\substack{\text { brown } \\(4 \text { moles })}}{4 \mathrm{NO}_2(\mathrm{~g})}+\underset{\text { colourless }}{\mathrm{O}_2(\mathrm{~g} \text { mole })}/latex]The rate of the reaction is the rate at which N2O5 decomposes or the rate at which O2 and NO2 are formed.
Here we assume that there is no change in the volume of the reaction system. In other words, there is no removal from or addition to the reaction system. As you can see, the number of moles of gases increases from 2 to 5, which means there is an increase in the pressure of the reaction system during the reaction.

The change in concentration of the reactant or the products can also be measured by measuring the increase in pressure.
Let us first study the formation of the product O2.
⇒ [latex]\text { Reaction rate }=\frac{\text { change in concentration of oxygen }}{\text { time interval }}\)
⇒ \(\frac{\Delta\left[\mathrm{O}_2\right]}{\Delta t}=\frac{\text { concentration of } \mathrm{O}_2 \text { at time } t_2-\text { concentration of } \mathrm{O}_2 \text { at time } t_1}{t_2-t_1}\)
Here square brackets indicate molar concentration. Shows the concentration as a function of time at 55°C for the reaction
⇒ \(2 \mathrm{~N}_2 \mathrm{O}_5(\mathrm{~g}) \rightarrow 4 \mathrm{NO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g})\)
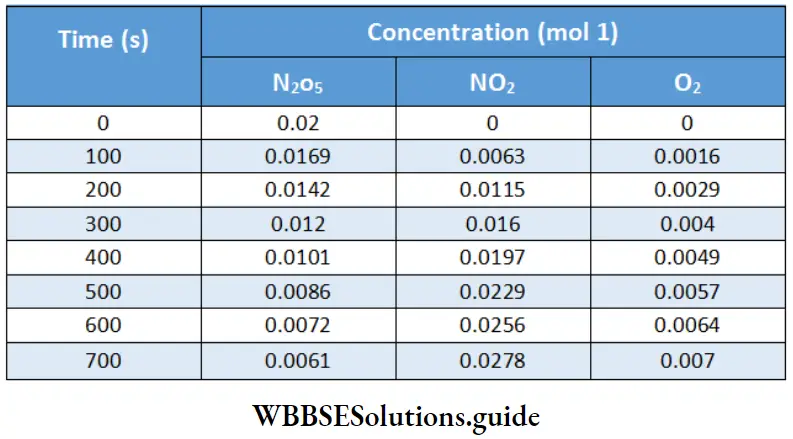
Refer to Table During the time period 400 s to 500 s, for example, the average rate of formation of O2 will be 8 x 10-6 mol L _1 s _1 because the rate of formation of
O2 = \(\frac{0.0057-0.0049}{500-400}=8 \times 10^{-6} \mathrm{~mol}^{-1} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
Now let us express the rate of the reaction in terms of the change in the concentration of NO2. During till same time period 400 seconds to 500 s, the rate of formation of NO2 will be
⇒\(\frac{0.0229-0.0197}{500-400}=3.2 \times 10^{-5} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
Rate of reaction and factors affecting chemical kinetics
The rate of formation of NO2 is 4 times that of O2 because for 2 mol of N2O5,1 mol of O2 and 4 mol of NC are formed. In other words, the rate is in accordance with the 4: 1 stoichiometric coefficient ratio of NO2 and C in the chemical equation.
Similarly, we can expect the rate of disappearance of N2O5 to be twice that of the formation of O2 As the products form, the reactant disappears. Hence, the value of A[N2O2 ]/Af turns out to be negative.
B all rates of reactions are reported as positive quantities. Thus, to maintain the reaction rate as a positive quantity the rate of a reaction is defined as the rate of formation of products or rate of disappearance of reactants divided by the corresponding stoichiometric coefficient in the balanced chemical equation, the coefficient being taken as positive for products and negative for reactants.
Thus, rate of disappearance of
⇒\(-\frac{[0.0086-0.0101]}{[500-400]}=1.5 \times 10^{-5} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1} \approx 2 \times 8 \times 10^{-6} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
Thus, the rate of formation of O2
⇒ \(\text { rate of formation of } \mathrm{NO}_2=\frac{1}{2} \text { the rate of decomposition of } \mathrm{N}_2 \mathrm{O}_5\)
or, \(\frac{\Delta\left[\mathrm{O}_2\right]}{\Delta t}=\frac{1}{4} \frac{\Delta\left[\mathrm{NO}_2\right]}{\Delta t}=-\frac{1}{2} \frac{\Delta\left[\mathrm{N}_2 \mathrm{O}_5\right]}{\Delta t}\)
Therefore, for the reaction
⇒ \(2 \mathrm{~N}_2 \mathrm{O}_5(\mathrm{~g}) \rightarrow 4 \mathrm{NO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g})\)
Rate of reaction = \(\frac{1}{2} \frac{\Delta\left[\mathrm{N}_2 \mathrm{O}_5\right]}{\Delta t}=\frac{1}{4} \frac{\Delta\left[\mathrm{NO}_2\right]}{\Delta t}=\frac{\Delta\left[\mathrm{O}_2\right]}{\Delta t}=8 \times 10^{-6} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
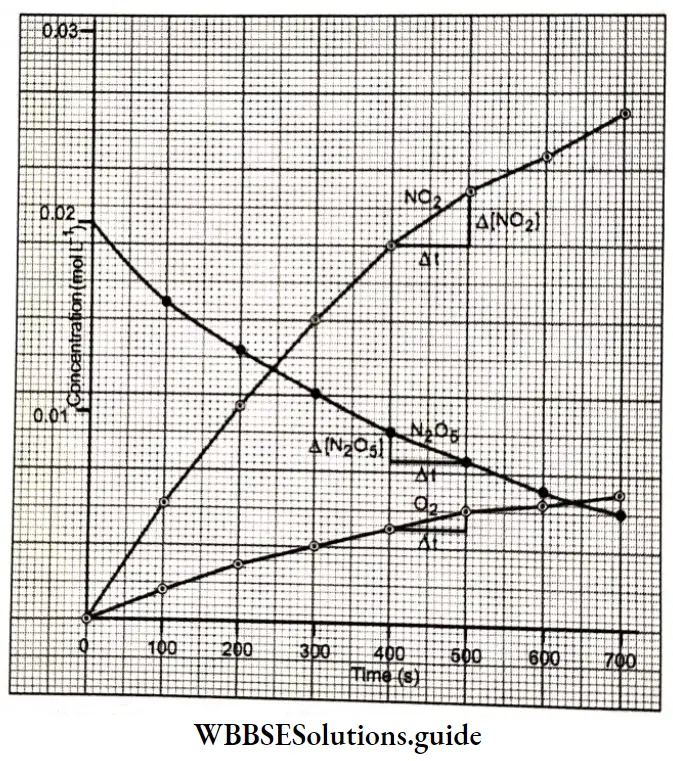
A plot of concentration versus time for the three gases NO2, N2O2, and O2 using the data from Table. As you can see, there are no straight lines, which means the rate (as determined by the slope of the curve) is different for different time intervals. The steeper the slope, the faster the rate. For example, the rate between 600 s and 700 s turns out to be 5.5 x 10-6 mol L-1s-1 while that between 300 s and 400 s is 9 x 106 mol L-1s-1.
The slope of the curve can be determined by finding the coordinates of any two points on the curve. If we choose the x coordinates of the points to be 600 s and 700 s, the respective y coordinates will be 0.0072 mol L-1 and 0.0061 mol L-1 (concentration of N2O2).
⇒ \(\text { Slope }=\frac{\text { change in } y}{\text { change in } x}=\frac{\Delta y}{\Delta x}=\frac{\Delta\left[\mathrm{N}_2 \mathrm{O}_5\right]}{\Delta t}\)
⇒ \(\frac{0.0061-0.0072}{700-600}=-1.1 \times 10^{-5} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
Rate = \(-\frac{1}{2} \cdot \frac{\Delta\left[\mathrm{N}_2 \mathrm{O}_5\right]}{\Delta t}=-\frac{1}{2} \times\left(-1.1 \times 10^{-5}\right)=5.5 \times 10^{-6} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
The rote of a reaction is not uniform during the course of the reaction, Initially, It la more but decreases with the decrease in the concentration of the reactant, Therefore, we may conclude Ilia! the mill rate (tetmnlHrit during n time internal to the accrue mission rule during that lime.
Chemical kinetics formulas and equations explained
Thus fin 10 A mol 1, 1 s 1the average reaction rate during the time interval 400 to 500 s, instantaneous rate If the change In concentration of the reactant and the product la determined consecutively at gradually decreasing time-intervals, a lime-interval which Is almost zero will be readied, The reaction rate determined at such an instant is called Instantaneous rale, l! is represented as (considering decomposition of nitrogen pentoxide)
⇒ \(r_{\text {lint }}=-\underset{\Delta t \rightarrow 0}{2} \frac{1}{\Delta t} \frac{\Delta\left[\mathrm{N}_2 \mathrm{O}_5\right]}{\Delta t}=-\frac{1}{2} \frac{d\left[\mathrm{~N}_2 \mathrm{O}_5\right]}{d t}=\frac{1}{4} \times \frac{d\left|\mathrm{NO}_2\right|}{d t}=\frac{d\left[\mathrm{O}_2\right]}{d t}\)
(note that the denominator contains the respective stoichiometric coefficients)
The instantaneous rate of the consumption of a reactant or formation of a product at some time / can be calculated by finding the slope of a graph of cells (reactant’s or product’s) molar concentrations plotted against the time, and the slope evaluated at the instant of interest.
The plot of the molar concentration of NO2 versus time. In order to find the Instantaneous rate of reaction at 400 s, a tangent Is drawn at this time and Its slope 11 is calculated as \(\left(\frac{\Delta y}{\Delta x}\right)\) as shown.
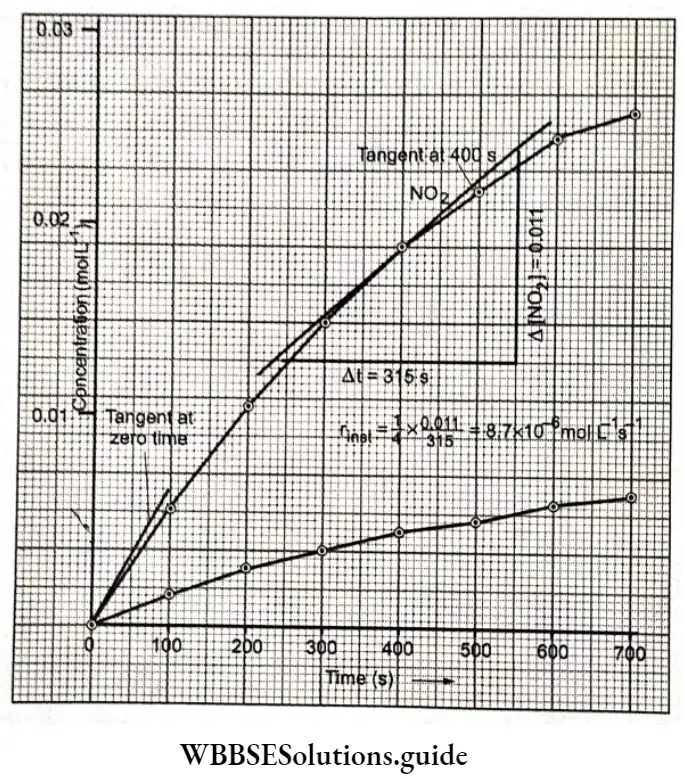
The instantaneous rate is then given by
⇒\(r_{\text {inst }}=\frac{1}{4} \text { slope }=\frac{1}{4} \times\left(\frac{0.011}{3.15}\right)=8.7 \times 10^{-6} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
We can also find the initial rate of the reaction, the rate when no products are formed, from the slope of the tangent at zero time
Initial rate = \(\frac{1}{4} \times\left(\frac{0.0038}{50}\right)=1.9 \times 10^{-5} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~s}^{-1}\)
It may be clearly seen that the instantaneous rate and initial rate are different.
Example 1. Express the relationship between the rate of production of iodine and the rate of consumption of hydrogen iodide in the following reaction.
⇒ \(2 \mathrm{HI} \rightarrow \mathrm{H}_2+\mathrm{I}_2\)
Solution:
The stoichiometries of I2 and HI in the reaction are 1 and 2 respectively.
Hence, the rate = \(-\frac{1}{2} \cdot \frac{\Delta[\mathrm{HI}]}{\Delta t}=\frac{\Delta\left[\mathrm{I}_2\right]}{\Delta t}\)
Now you know that the rate of a reaction is determined by measuring the change in concentration of any of the reactants or the products. But how is the concentration or a change in it during the course of the reaction measured? As you know, whether or not a reaction has occurred can be determined by observing changes in any of the reactants or products, such as changes in state, temperature, and color. Thus, by observing any such change in the property of either a reactant or a product, the change in its concentration can be determined.
Titration, colorimetry (a technique in which the absorption of light by a substance is measured), conductimetry (measuring the conductance of a solution) and pressure measurements are some of the techniques used to follow the concentration change of reactants or products.
Factors Affecting The Rate Of A Reaction
As you can see the rate of a reaction decreases with an increase in time. The rates tend to decrease as the reaction proceeds forward and reactants are consumed. You know that reactions take place as a result of collisions between molecules of reactants.
According to the kinetic molecular theory, the pressure exerted by a gas is proportional to the frequency with which molecules of the gas collide with tire walls of the container. The more the number of molecules present in a given volume, the greater is the number of collisions.
And if the number of collisions between the reacting molecules is larger (at the beginning of the reaction), the higher will be the rate.of the reaction. As the concentration of reactants decreases, the rate decreases.
NCERT class 12 chemical kinetics chapter notes
Apart from the concentration of the reactants, there are several factors which influence the rate of a reaction. These are the surface area and concentration of the reactants, temperature and pressure conditions of the reaction, and the presence of catalysts and light. Electric and magnetic fields also affect the rate of a reaction.
A larger surface area helps in better contact between the reactants and hence leads to an increased number of collisions between the reactant molecules resulting in a faster rate of reaction. Increasing the concentration of a reactant does not always increase the rate of a reaction.
In general, increasing the temperature increases the rate of chemical reactions. This is why food, if not refrigerated, spoils faster in summer than in winter.
Influence Of Concentration Rate Law
You know that the rate of a reaction at a given temperature depends on the concentration of one or more reactants and products.
Let us consider the reaction between bromine and formic acid in an aqueous solution catalyzed by acid.
⇒ \(\mathrm{Br}_2(\mathrm{aq})+\mathrm{HCOOH}(\mathrm{aq}) \stackrel{\mathrm{H}^*}{\longrightarrow} 2 \mathrm{Br}^{-}(\mathrm{aq})+2 \mathrm{H}^{+}(\mathrm{aq})+\mathrm{CO}_2(\mathrm{~g})\)
When the concentration of bromine during the course of the reaction is plotted against time, we obtain a curve as shown. The rate of this reaction can thus be expressed as
⇒ \(\text { rate }=-\frac{d\left[\mathrm{Br}_2\right]}{d t}\)
A plot of the reaction rate against the concentration of bromine is shown. It is a straig] line, indicating that the reaction rate is directly proportional to the concentration of bromine, i.e.„
reaction rate ac [Br2 ]
or, reaction rate = k[Br2].
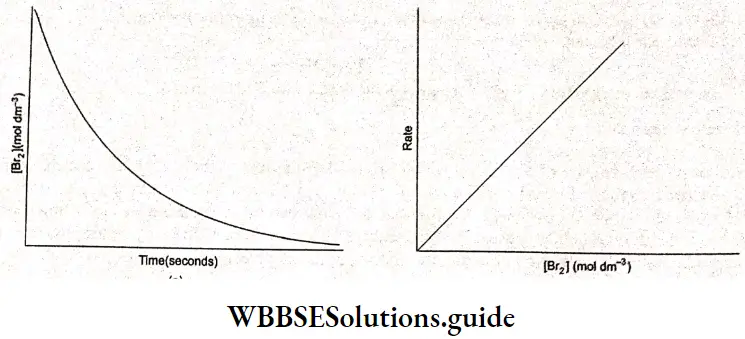
Here k is called the rate constant or velocity constant and is characteristic of the reaction being studied. The rate constant is independent of the concentrations of the reactants. However, it depends on the temperature. An equation of this kind which is experimentally determined for a reaction is called the rate equation or rate law of that equation. The rate law of a reaction gives the dependence of the reaction rate on the concentration of each reactant.
Experimental findings reveal that the rate of a reaction is usually proportional to the molar concentrations of the reactants raised to a simple power.
⇒ \(\text { Rate }=k[R]^n\)
In the given equation, [R] is the molar concentration of the reactant raised to a simple power n, which may or may not be the same as the stoichiometric coefficient of the reactants in the balanced chemical equation.
Similarly, for a general reaction
mA + nB → product
the rate law is of the form
⇒ \(\text { rate }=-\frac{d[\mathrm{~A}]}{d t}=k[\mathrm{~A}]^m[\mathrm{~B}]^n\)
This equation is called the differential rate equation or rate expression. The exponents m and n are usually positive integers. These exponents for a reaction are determined experimentally as also the rate constant k.
The rate law for the reaction between Br2 and formic acid turns out to be
⇒ \(\text { rate }=k\left[\mathrm{Br}_2\right][\mathrm{HCOOH}]^0\)
or, \(\text { rate }=k\left[\mathrm{Br}_2\right]\)
In this case, the two exponents m and n are 1 and 0 respectively. The rate, therefore, depends only on the concentration of Br2 and not on that of HCOOH. We could either have all the reactants appearing in the rate law or only some may appear; the exponents may or may not be the same as the stoichiometric coefficients.
Reaction order:
Let us now see how the rate is affected by the value of the exponent. In Equation 2, if m =1, it means that the rate of the reaction depends linearly on the concentration of one of the reactants, A. If the concentration of A is doubled so will be the rate. But if it is assumed that m = 2, then if the concentration of A is doubled, the rate quadruples, i.e., increases four times (∵ [2A] = 4[A]2)
Similarly, the rate of the reaction is also affected by the value of n. If the concentration of A is kept constant and that of B is doubled, and the rate also doubles, it means the rate of the reaction depends linearly on the concentration of B. Now in the case of the reactant A, if we take the second assumption (m¯²) to be true then the rate equation for the reaction will be
rate = k [A]²[B]¹
=k[A]²[B].
Thus, exponents m and n in the rate law indicate how sensitive the rate is to changes in the concentration of A and B. The dependence of the reaction rate on concentration is expressed in terms of reaction order.
For the given chemical reaction the order of the reaction with respect to A is m (the exponent of A in the rate law) and the order with respect to B is n. The overall order of a reaction is the sum of the exponents in the rate law and is m + n for the above reaction.
Definition and types of rate of reaction in chemical kinetics
A reaction whose overall order is 1 is called a first-order reaction, one with an order of 2 is called a second-order reaction, one with order 3 is called a third-order reaction, and so on.
For example, the reaction involving the decomposition of hydrogen peroxide is
⇒ \(2 \mathrm{H}_2 \mathrm{O}_2(\mathrm{aq}) \rightarrow 2 \mathrm{H}_2 \mathrm{O}(\mathrm{l})+\mathrm{O}_2(\mathrm{~g})\)
The rate law for this reaction is
rate = k[H2O2].
Its order with respect to H2O22 is one and its overall order is also one. Hence it is a first-order reaction.
Let us consider the reaction between HCl and NaOH
⇒ \(\mathrm{Na}^{+}+\mathrm{Cl}^{-}+\mathrm{H}_3 \mathrm{O}^{+}+\mathrm{OH}^{-} \rightarrow \mathrm{H}_2 \mathrm{O}+\mathrm{Na}^{+}+\mathrm{Cl}^{-}\)
It is of the first order with respect to each of the reactants H3O+ and OH–.
⇒ \(\text { Rate }=k\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{OH}^{-}\right]\)
But the overall order is 2. Hence, it is a second-order reaction.
NO2 (g) + CO(g) → NO(g) + CO2 (g)
The rate law of the given reaction is
rate = k [NO2]²[CO]O2.
The order of this reaction is two with respect to NO2 and zero with respect to CO. The overall order is two because [CO]0 =1, just as in algebra. It is called a second-order reaction. The concentration of carbon monoxide raised to power zero implies that the rate of the reaction is independent of the concentration of CO provided that some CO is present.
H2(g) + 2NO(g) → N2O(g) + H2O(g)
The rate law for this reaction is
rate = k [NO]2[H2 ]
The order of the reaction is two with respect to NO and one with respect to H2. The overall order of the reaction is three. Therefore it is a third-order reaction.
Note that the order of a reaction or the power of the concentration of the reactant in the rate expression of a reaction has no connection with the stoichiometric coefficient of the reactant.
It is also important to note that a rate law is established experimentally and cannot in general be inferred from the chemical equation for the reaction.
Unit of rate constant You know that
⇒ \(k=\frac{\text { rate }}{{\text { (concentration })^n}^{(}}\)
The SI unit of concentration is mol L-1 and that of time is s.
Therefore,
⇒ \(k=\frac{\mathrm{mol} \mathrm{L}^{-1}}{\mathrm{~s}} \times \frac{1}{(\text { concentration })^n}\)
Thus, the units of k for different reaction orders will be as follows.

Integrated Rate Laws
As discussed earlier in the chapter, the rate law of a reaction gives the rate of a reaction at a given instant. In other words, it tells us the rate of a reaction at a certain composition of the reaction mixture. If we have to find out how the concentrations depend on time, we need to integrate the differential rate expressions.
An integrated rate law is an expression that gives the concentration of a species as a function of time. Another way of saying this is that the integrated rate law is used to predict the concentration of a species at any time after the start of the reaction.
Also, the law helps find the rate constant and thereby the order of the reaction. Let us derive and study the rate laws for zero- and first-order reactions.
Zero-order reactions:
Consider the reaction A → P, which obeys the zero-order rate law, i.e., the rate of the reaction is proportional to the concentration of the reactants, raised to the power zero.
⇒ \(\text { Rate }=-\frac{d[\mathrm{~A}]}{d t}=k[\mathrm{~A}]^0=k[1]=k\)
As the equation shows, the rate of such a reaction is independent of the concentration of the reactant throughout the course of the reaction. Integrating Equation 4.1 we get
⇒ \(\int_{[\hat{\mathrm{A}}]_0}^{[\mathrm{A}]} d \mathrm{~A}=-\int_0^t k d t\)
⇒ \([\mathrm{A}]-[\mathrm{A}]_0=-k t\)
or \([\mathrm{A}]=-k t+[\mathrm{A}]_0\)
where [A]0 is the initial concentration of A and [A] is its final concentration.
This equation is called the integrated rate law.
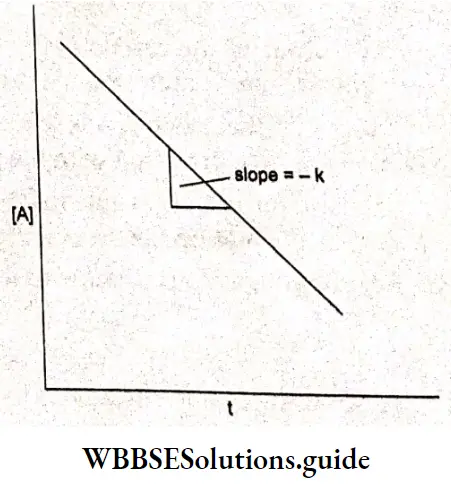
This is an equation of the form y = mx + c, which is the equation of a straight line. Hence a graph of [A] vs time is a straight line with a slope, -k. Zero-order reactions are uncommon but they can occur under special conditions. For example, the decomposition of gaseous ammonia on a hot platinum surface is a zero-order reaction.
⇒ \(2 \mathrm{NH}_3(\mathrm{~g}) \stackrel{\mathrm{Pt}, 1130 \mathrm{~K}}{\longrightarrow} \mathrm{N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g})\)
⇒ \(\text { Rate }=k\left[\mathrm{NH}_3\right]^0=k\)
First-order reactions:
For a first-order reaction of the type
A → P
the differential rate law is
⇒ \(\text { rate }=-\frac{d[\mathrm{~A}]}{d t}=k[\mathrm{~A}]\)
⇒ \(-\frac{d[\mathrm{~A}]}{[\mathrm{A}]}=k d t .\)
On integrating from f = 0, when the concentration of A is [A]0, to the time t, when the molar concentration of A is [A], we obtain
⇒ \(|-\ln [\mathrm{A}]|_{[\mathrm{A}]_0}^{[\mathrm{A}]}=k|t|_0^t\)
⇒ \(-\ln [\mathrm{A}]+\ln [\mathrm{A}]_0=k t\)
or, \(\ln [\mathrm{A}]=\ln [\mathrm{A}]_0-k t\)
The exponential form of the equation can be given as
⇒ \(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=-k t\)
This suggests that for all first-order reactions the concentration of the reactant decays exponentially with time. [A] is the concentration of A at time f and [A]0 is its initial concentration. Thus, the integrated rate law equation can be written as
⇒ \(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=-k t\)
The above equation can also be written as
⇒ \(k=\frac{1}{t} \ln \frac{[\mathrm{A}]_0}{[\mathrm{~A}]}\)
or, \(k=\frac{1}{\left(t_2-t_1\right)} \ln \frac{\left[\mathrm{A}_1\right]}{\left[\mathrm{A}_2\right]}\)
Here the concentrations [A x ] and [A 2 ] are at times fx and 12 respectively.
Integrated rate laws can be used to determine the order and rate constant of a reaction. For this we need experimental data of the concentration of the reactant as a function of time.
For a first-order reaction, the plot of In [A] vs f should be a straight line. This is clear from the equation
In [A] = In [A]0– kt
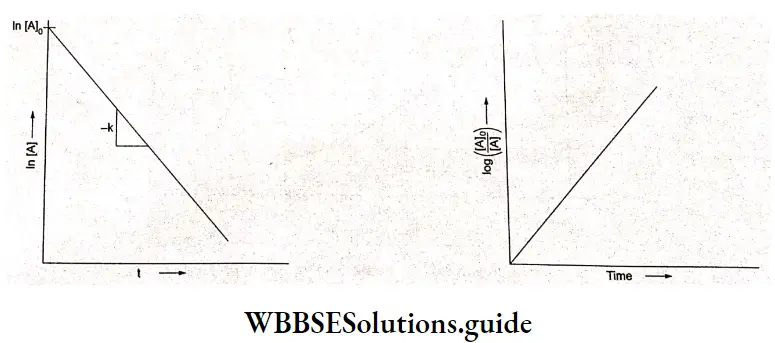
It is of the form Y-a~ bX. where Y is In [A], X is I. b, the slope, is(-) and the intercept is In[A]0.
For any reaction, we plot In [A] vs. f from the experimental data of that reaction, and if it does not give a straight line, the reaction is not of the first order.
Equation 43 may also be written as
⇒ \(2.303 \log \left\{\frac{[\mathrm{A}]}{[\mathrm{A}]_0}\right\}=-k t\)
or, \(-2.303 \log \left\{\frac{[\mathrm{A}]}{[\mathrm{A}]_0}\right\}=k t\)
or, \(2.303 \log \left\{\frac{[\mathrm{A}]_0}{[\mathrm{~A}]}\right\}=k t\)
or, \(\log \left\{\frac{[\mathrm{A}]_0}{[\mathrm{~A}]}\right\}=\frac{k t}{2.303}\)
A plot of log \(\left\{\frac{[\mathrm{A}]_0}{[\mathrm{~A}]}\right\}\) versus t gives a straight line with slope \(\frac{k}{2.303}\)
The thermal decomposition of N2 Og dealt with at the beginning of this chapter is a first-order reaction and its rate law is rate = k[N205].
The decomposition of hydrogen peroxide in a dilute sodium hydroxide solution is described by the equation
2H2O2(aq) → 2H2O(1) + O2 (g)
Let us find the order of this reaction. The concentration of H2O2 as a function of time was found to be as follows.
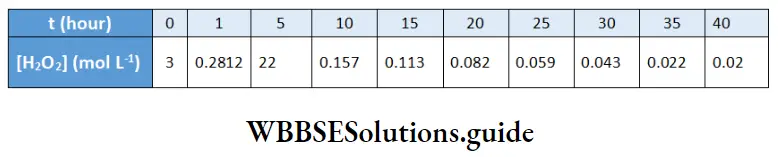
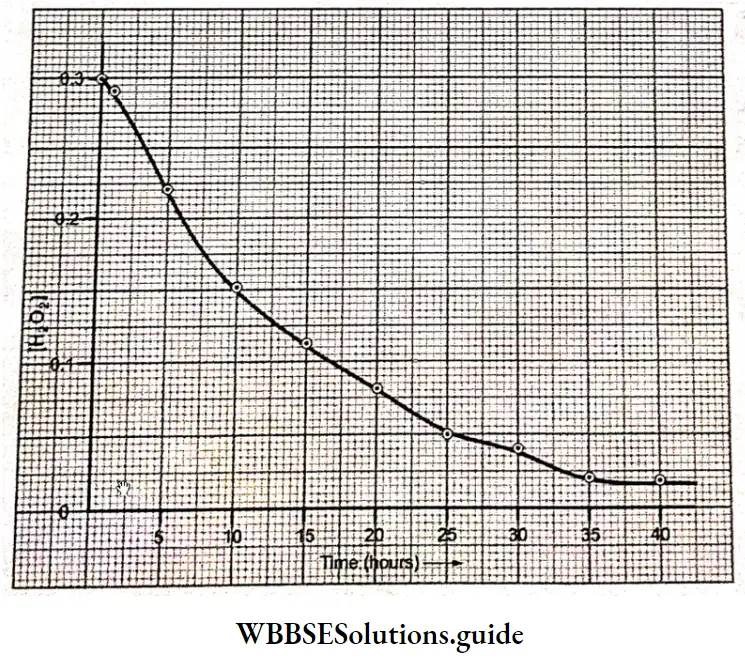
A plot of the concentration of H2O2 as a function of time. It may be seen from the graph the concentration of the reactant decays exponentially with time. On plotting In[H2O2] versus time, a fight line is obtained indicating that the reaction is of the first order
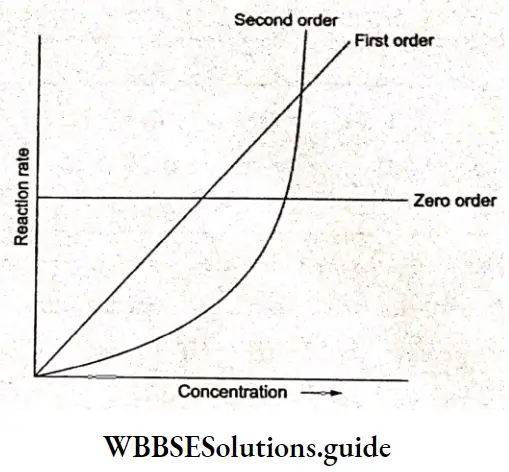
Whether a reaction is of zero, first, or second order can be found by a simple plot of the reaction rate concentration of the reactant.
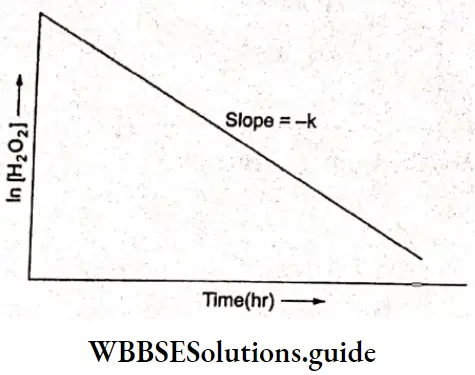
Example 1. The reaction of N2O2 with water produces HNO3.
⇒ \(\mathrm{N}_2 \mathrm{O}_5+\mathrm{H}_2 \mathrm{O} \rightarrow 2 \mathrm{HNO}_3\)
The reaction is of the first order with respect to each reactant. Write the rate law for the reaction.
When \(\left[\mathrm{N}_2 \mathrm{O}_5\right] \text { is } 0.13 \times 10^{-6} \mathrm{~mol} \mathrm{~L}^{-1}\) and \(\left[\mathrm{H}_2 \mathrm{O}\right]=2.3 \times 10^{-4} \mathrm{~mol} \mathrm{~L}^{-1}\)
the rate of the reaction is 4.55 x 10-10 mol L-1 min-1. What is the rate constant for the reaction?
Solution:
Since the reaction is of the first order with respect to each reactant, the power of each reactant must be 1 in the rate law.
Therefore, rate = k[N2O5 ][H2O].
Given that rate = \(4.55 \times 10^{-10} \mathrm{~mol} \mathrm{~L}^{-1} \mathrm{~min}^{-1},\left[\mathrm{~N}_2 \mathrm{O}_5\right]=0.13 \times 10^{-6} \mathrm{~mol} \mathrm{~L}^{-1}\)
and \(\left[\mathrm{H}_2 \mathrm{O}\right]=2.3 \times 10^{-4} \mathrm{~mol} \mathrm{~L}^{-1}\)
Substituting these values in the rate law
⇒ \(4.55 \times 10^{-10}=k\left(0.13 \times 10^{-6}\right)\left(2.3 \times 10^{-4}\right)\)
or, \(k=\frac{4.55 \times 10^{-10}}{0.13 \times 10^{-6} \times 2.3 \times 10^{-4}}=15.05 \mathrm{~L} \mathrm{~mol}^{-1} \mathrm{~s}^{-1}\)
The rate constant is 15.05 L mol-1 s-1.
Example 2. The rate of formation of a dimer in a second-order dimerization reaction is 9.1 x 10-6 mol L-1 s-1 at 0.05 M monomer concentration. Calculate the rate constant.
Solution:
Given
The rate of formation of a dimer in a second-order dimerization reaction is 9.1 x 10-6 mol L-1 s-1 at 0.05 M monomer concentration.
For a second-order reaction
rate = k[A]²
or 9.1 x106 =k(0.05)²
⇒ \(k=\frac{9.1 \times 10^{-6}}{(0.05)^2}=3.64 \times 10^{-3} \mathrm{~L} \mathrm{~mol}^{-1} \mathrm{~s}^{-1}\)
Example 3. Consider the following data the reaction.
A + B → product
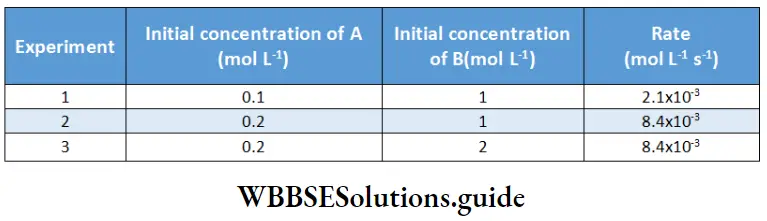
Determine the order of reaction with respect to A and B, and the overall order of the reaction,
Solution:
In experiments 1 and 2, the concentration of B is constant. Hence,
⇒ \(\frac{\text { rate }_1}{\text { rate }_2}=\frac{k[\mathrm{~A}]_1^a[\mathrm{~B}]_1^b}{k[\mathrm{~A}]_2^a[\mathrm{~B}]_2^b}=\frac{k[\mathrm{~A}]_1^a}{k[\mathrm{~A}]_2^a}\)
⇒ \(\log \text { rate }_1-\log \text { rate }_2=a \log \frac{[\mathrm{A}]_1}{[\mathrm{~A}]_2}\)
On substituting the respective values, we get
⇒ \(\log 2.1 \times 10^{-3}-\log 8.4 \times 10^{-3}=a \log \left(\frac{0.1}{0.2}\right)=a \log (0.5)\)
or, \(-2.6778-(-2.0758)=a(-0.3010)\)
or, \(-0.6021=a(-0.3010)\)
or, \(\frac{0.6021}{0.3010}=a\)
⇒ a = 2.
The order with respect to A is 2.
Now consider experiments 2 and 3, where the concentration of A is the same. We get
⇒ \(\log \text { rate }_2-\log \text { rate }_3=b \log \left(\frac{[\mathrm{B}]_2}{[\mathrm{~B}]_3}\right)\)
or, \(\log 8.4 \times 10^{-3}-\log 8.4 \times 10^{-3}=b \log \left(\frac{1}{2}\right)\)
b log (0.5) = 0
⇒ b = 0 [as log (0.5) ≠ 0].
The order with respect to B is zero. Hence the overall order is a + b = 2.
Example 4. The following data was obtained at 300 K for the reaction 2A + B → C+ D.
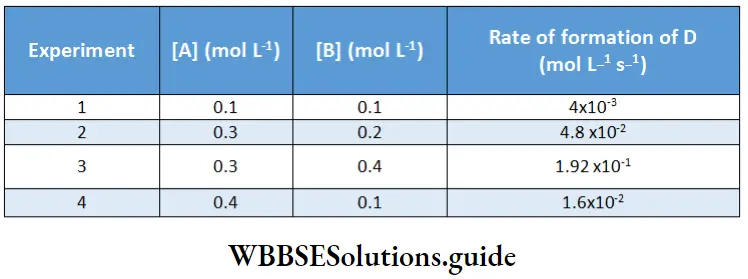
Calculate the rate of formation of D when [A] = 0.7 mol L-1 and[B] = 0.3 mol L-1.
Solution:
In experiments 1 and 4, the concentration of B is the same.
Therefore, \(\log \frac{\text { rate }_1}{\text { rate }_4}=a \log \frac{[\mathrm{A}]_1}{[\mathrm{~A}]_4}\)
⇒ \(\log \frac{4 \times 10^{-3}}{1.6 \times 10^{-2}}=a \log \left(\frac{0.1}{0.4}\right)\)
log 0.25 = a log (0.25)
a = 1.
The order with respect to A is one.
In experiments 2 and 3, the concentration of A is the same. Therefore,
⇒ \(\log \frac{\text { rate }_2}{\text { rate }_3}=b \log \frac{[\mathrm{B}]_2}{[\mathrm{~B}]_3}\)
or, \(\log \frac{4.8 \times 10^{-2}}{1.92 \times 10^{-1}}=b \log \left(\frac{0.2}{0.4}\right)\)
or log 0.25 = blog (0.5)
⇒ -0.6021 = b(-0.3010)
⇒ b = 2.
The order with respect to B is two.
Hence, the overall order of the reaction is 3. The rate law for the reaction is
rate = k[A][B]².
Taking the observed values from any one of the experiments (say 1) and substituting the rate and concentration of A and B, we get
4 x 10¯³ = k(0.1)(0.1)²
⇒ \(k(0.1)(0.1)^2=4 \times 10^{-3} \Rightarrow k=\frac{4 \times 10^{-3}}{1 \times 10^{-3}}=4 \mathrm{~L}^2 \mathrm{~mol}^{-2} \mathrm{~min}\)
The value of the rate can now be determined with A = 0.7 mol L-1 and B = 0.3 mol L-1 by simply substituting the values of the concentrations and k in the rate law.
Rate = 4 x (0.7)(0.3)2 = (2.8)(0.09).
∴ rate = 0.252 mol L¯¹ min¯¹
Example 5. For a reaction A+B → C the following data was obtained.
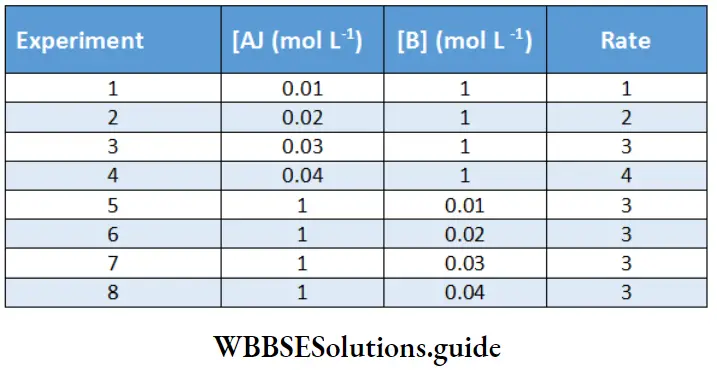
By plotting a graph of reaction rate vs. concentration of the reactant, find the order of the reaction.
Solution:
In experiments 1 2,3 and 4 the concentration of B is much more than that of A. Hence, these data can tv used to determine order with respect to A,
⇒ \(\text { Rate }=K \mid A]^a[B]^b\)
⇒ \(\text { Nate }=K(A)^2\)
Since the plot of rate vs. concentration of A is a straight line, the reaction is of the first order with respect to A. In experiments 5, 6. 7, and 8 the concentration of A is in excess and hence these data can be used to determine the order with respect to B. A plot of rate vs. concentration of B is obtained as given below on the right.
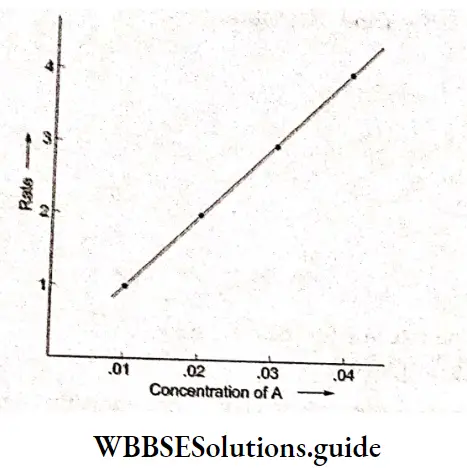
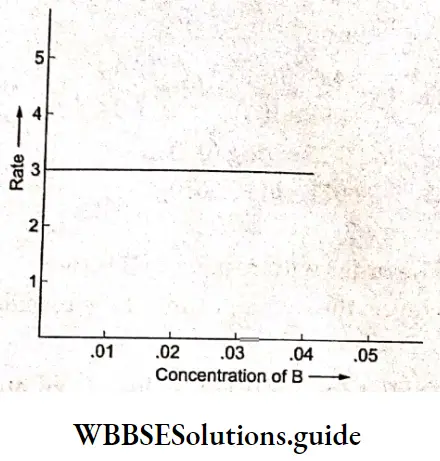
Since we obtain a line parallel to the x-axis, the rate is independent of the concentration of B. The reaction is of zero order with respect to B. Even without plotting the graph, a close look at the data tells us that the rate is constant for all experiments 4-8 even though the concentration of B is varying. Hence the order is zero with respect to B. The overall order of the reaction is 1 + 0 =1.
Example 6. The reaction between NO and H2 occurs as follows.
2H2(g) + 2NO(g)4 – 2H2O(g) + N2(g)
Several experiments were performed by keeping the concentration of one of the reactants constant and changing the concentration of the other. The initial rates obtained are tabulated below.
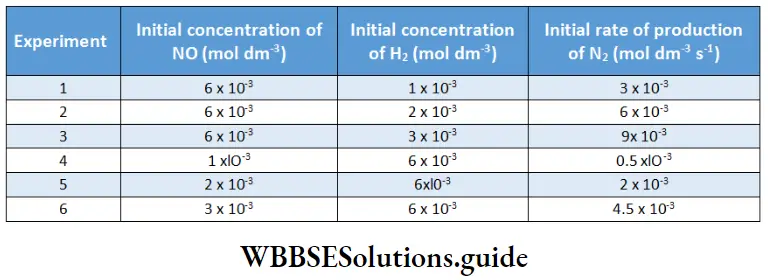
What is the order of the reaction with respect to NO and H2? Write down the rate law. Calculate the rate constant k.
Solution:
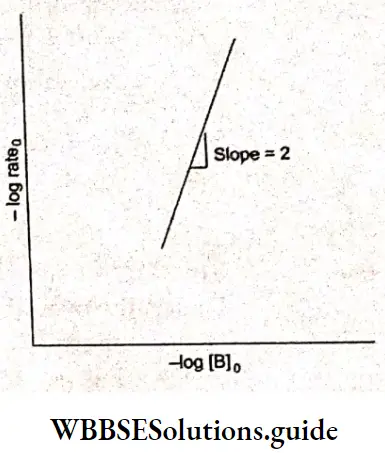
In experiments 1, 2, and 3 the concentration of NO is constant; the data from these experiments give us the order of the reaction with respect to H2. (The symbol 0 in the subscript denotes the initial conditions.)
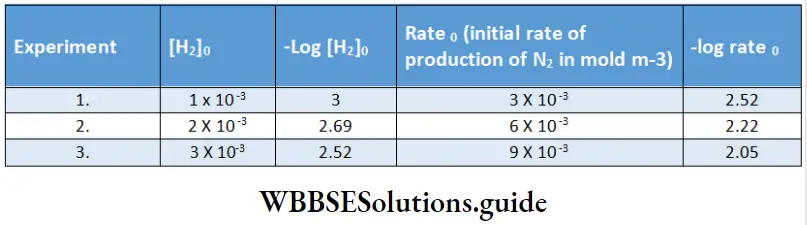
The plot of -log rate 0 vs -log [H2]0 is a straight line with slope 1.
Hence, the order with respect to H2 is one. Similarly, we can plot -log rate O vs -log[NO]0 for experiments 4, 5, and 6.
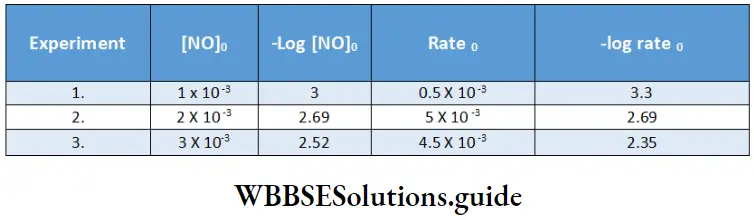
The slope obtained from the plot gives a value of 2, which is the order with respect to NO.
Hence, the overall order = order (H2) + order (NO)
=1 + 2
= 3.
Hence, the reaction is of the third order.
The rate law is rate = k[H2][NO]2.
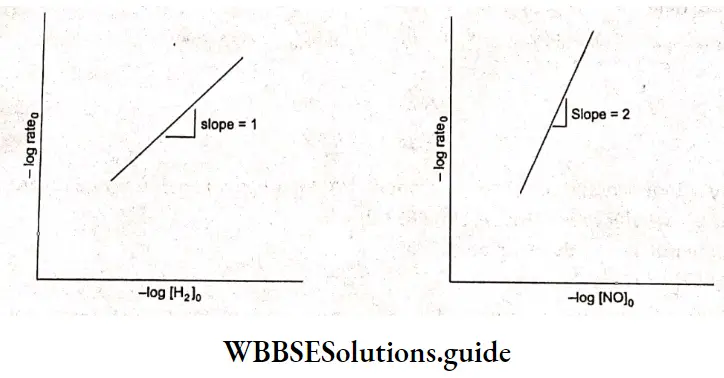
Taking the observed values from any one of the experiments and substituting the concentration of H2 and NO, we get (experiment 1)
⇒ \(3 \times 10^{-3}=k \cdot\left(1 \times 10^{-3}\right)\left(6 \times 10^{-3}\right)^2\)
⇒ \(k=\frac{3 \times 10^{-3}}{6 \times 10^{-9}}=0.5 \times 10^6=5 \times 10^5 \mathrm{dm}^6 \mathrm{~mol}^{-2} \mathrm{~s}^{-1}\)
Example 7. The reaction A+ B → C was studied by performing several experiments and the following information was obtained.
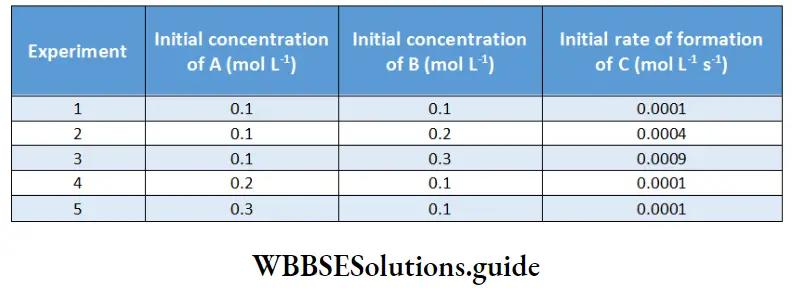
What is the order of the reaction with respect to A anti that with respect to B Write down the rate law.
Calculate the rate constant, k.
Solution:
In experiments 1, 2, and 3, the concentration of A is constant; hence these data can give us the order with respect to B.
In the initial rate method, \(-\log \text { rate }_0 \text { vs }-\log [\mathrm{B}]_0\) is plotted
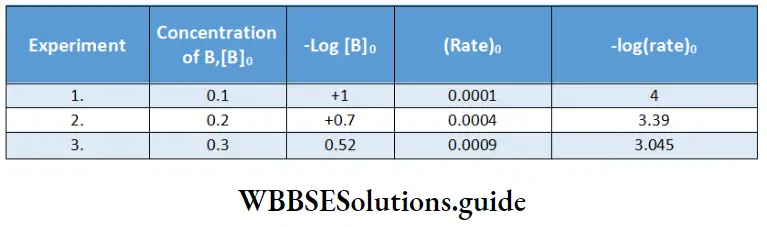
The slope can be determined from the two corresponding values on the x- and y-axes.
For instance,
⇒ \(\frac{y_2-y_1}{x_2-x_1}=\frac{4-3.39}{1-0.7} \cong 2\).
Therefore the order with respect to B is two.
Using the values obtained from experiments 4 and 5, we get the following data.
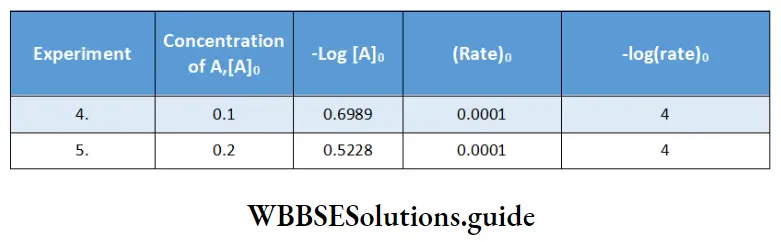
The rate is independent of the concentration of A. Hence, the reaction is of zero order in A.
Therefore, the overall order of the reaction (2 + 0) = 2.
Hence, the rate law of the reaction,
rate = k[A]°[B]².
On substituting the values obtained from experiment 1 in the rate law equation, we calculate the
rate constant.
0.0001 = k[0.1][0.1]²
⇒ \(\frac{0.0001}{(0.1)^3}=k\)
k- 0.1 L mol-1 s-1
Example 8. A first-order reaction is 30% complete in one hour. Calculate the rate constant for the reaction. In how much time will the reaction proceed to 80% completion?
Solution:
Given
A first-order reaction is 30% complete in one hour. Calculate the rate constant for the reaction.
For a first-order reaction, the integrated rate law is
⇒ \(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=-k t\)
If the reaction proceeds to 30% completion, it means 30% of [A] has been consumed and 70% [A] remains.
Substituting 100 for [A]0 70 for [A] and 60 min for t, we get
⇒ \(\ln \left(\frac{70}{100}\right)=-k \times 60\)
or, 2.303 log (0.7)= -k x 60
or, \(k=\frac{\log (0.7)}{60}=5.95 \times 10^{-3} \mathrm{~min}^{-1}\)
For the reaction to proceed to 80% completion, A must become 20.
In \(\ln \left(\frac{20}{100}\right)=-5.95 \times 10^{-3} \times t\)
or, \(t=\frac{-2.303 \log (0.2)}{1.12 \times 10^{-3}}=270.5 \mathrm{~min}\)
Example 9. A reaction that is of the first order with respect to reactant A has a rate constant of 5 min-1. If we start with 5 mol L-1 of A, would the concentration of A reach 0.05 mol L-1?
Solution:
Given
A reaction that is of the first order with respect to reactant A has a rate constant of 5 min-1. If we start with 5 mol L-1 of A,
For a first-order reaction
⇒ \(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=-k t\),
where [A] is the concentration of A at time t and [A]0 is the initial concentration of A.
Substituting the values of [A] = 0.05 mol L-1, [A]0 =5 mol L-1and k =5 min-1, we get
⇒ \(\ln \frac{0.05}{5}=-5 t\)
or, \(-\frac{1}{5} \ln \frac{0.05}{5}=t\)
⇒ \(-\frac{1}{5} \times 2.303 \log 0.01=t\)
⇒ \(-0.4606 \log \left(1 \times 10^{-2}\right)=t\)
⇒ -0.4606 x (-2) =t
⇒ 0.9212 = t
∴ t= 0.9212 min or 55.2 s.
It takes 55.2 seconds for the concentration of A to reach 0.05 mol L-1.
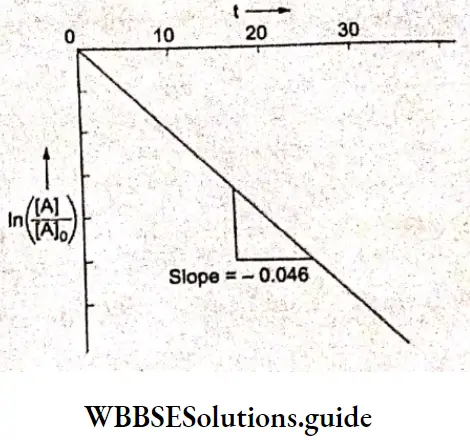
Example 10. For the isomerization of cyclopropanone to propene in the gaseous phase at 433°C, the following data were obtained.
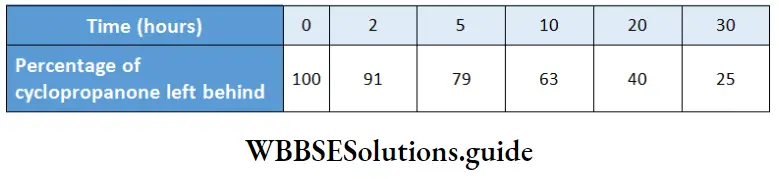
Find the order of the reaction and calculate the rate constant.
Solution:
If it is a first-order reaction, a plot of In \(\frac{[\mathrm{A}]}{[\mathrm{A}]_0}\) vs. t should be a straight line. Considering [A]0 =100 and the other values at various times as [A], we can calculate In \(\left(\frac{[\mathrm{A}]}{[\mathrm{A}]_0}\right)\)

A plot of In \(\frac{[\mathrm{A}]}{[\mathrm{A}]_0}\) vs f is a straight line with slope = -0.046
Hence it is n first-order reaction.
Since slope = -k,
k = 0.046 hours-1
⇒ \(\frac{0.046}{3600}=1.277 \times 10^{-5} \mathrm{~s}^{-1}\)
Example 11. The rate of decomposition of hydrogen peroxide at a particular temperature was measured by titrating its solution acidic KMn04 solution. The following results were obtained
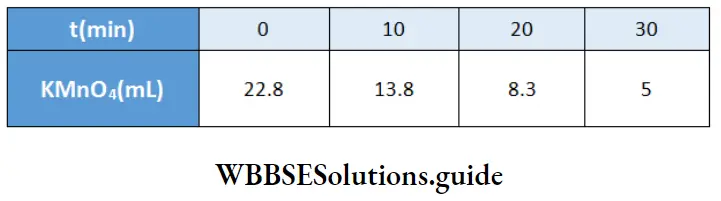
Show that it is a first-order reaction. Calculate the rate constant of the reaction.
Solution:
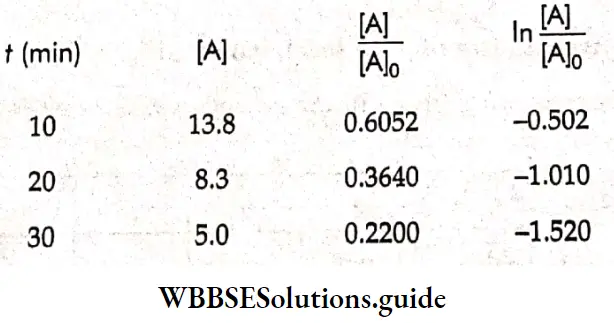
The titer value of KMnO4 is proportionate to the amount of H2O2 left undecomposed.
For a first-order reaction,
In \(\frac{[\mathrm{A}]}{[\mathrm{A}]_0}\) = -kt and a plot of in \(\frac{[\mathrm{A}]}{[\mathrm{A}]_0}\) vs t is a straight line.
[A]0 = 22.8 (at time = 0 min).
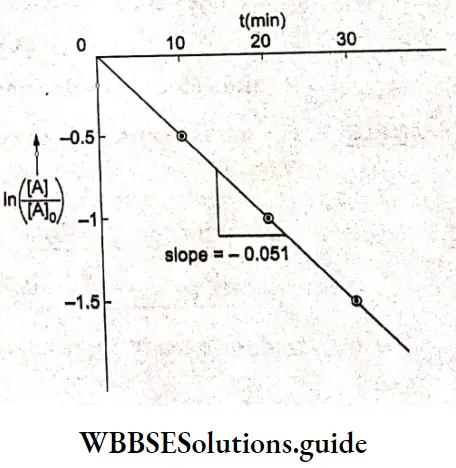
Since the plot is a straight line, the reaction is of the first order.
And slope = -0.051 = -k
=> k= 0.051 min-1.
Example 12. Consider the decomposition of SO2Cl2 to SO2 and Cl2. It follows first-order kinetics at 675 K. If the rate constant is 2×10-5 min-1, find the percentage of SO2Cl2 that decomposes in 57.7 hours.
Solution:
Given
Consider the decomposition of SO2Cl2 to SO2 and Cl2. It follows first-order kinetics at 675 K. If the rate constant is 2×10-5 min-1,
For a first-order reaction
⇒ \(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=-k t ; \text { let } \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=\mathrm{X}\)
Then, In \((X)=-2 \times 10^{-5} \times 57.7 \times 60\)
⇒ \(\log X=-\frac{0.06924}{2.303}=-0.0300\)
X = 0.9332.
The ratio of SO2Cl2 remaining undissociated to the initial amount is 0.9332.
0.93 x 100= 93% of SO2Cl2 remains undissociated.
This means 100- 93 = 7% of SO2Cl2 would have dissociated in 57.7 hours.
Factors affecting the rate of chemical reaction
Example 13. The dehydration of oxalic acid occurs according to the equation
H2C2O4 → CO + CO2 + H2O
The reaction is followed by titrating oxalic acid With KMnO4. In one such experiment, the following data were obtained.
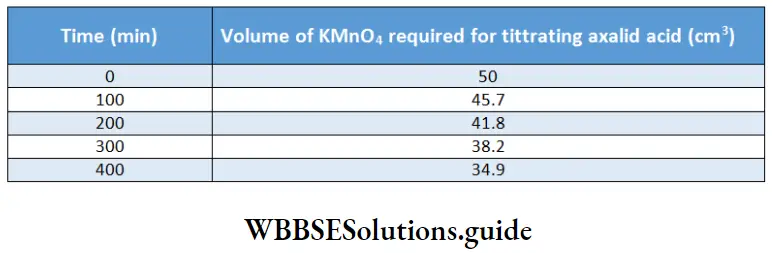
Find the order of the reaction and the rate constant.
Solution:
Here, we are titrating the oxalic acid that remains undissociated. Hence, the litre value at zero time gives the initial concentration of oxalic acid and other titer values indicate the amount of oxalic acid left behind.
For a first-order reaction a plot of In [A] vs. t must be a straight line.
In [A] = In [oxalic acid remaining undissociated].
Since we do not know the tire concentration of oxalic acid, the ratio of the titer values can give us the ratio of concentrations. At time 0 min, tire initial concentration, [A]0 = 50.

The value of k can be determined from the equation
⇒ \(k=-\left(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}\right) \times \frac{1}{t}\)
Since the value of k is constant, the reaction is of the first order.
Half-life
The half-life of a reactant, denoted by f1/2, is the time taken for the concentration of a reactant to fall to half of it initial amount.
For a first-order reaction, we can find the half-life of reactant A by substituting
⇒ \([\mathrm{A}]=\frac{1}{2}[\mathrm{~A}]_0 \text { and } t=t_{1 / 2}\)
Equation 4.3, so that
⇒ \(\ln \left(\frac{\frac{1}{2}[\mathrm{~A}]_0}{[\mathrm{~A}]_0}\right)=-k t_{1 / 2}\)
⇒ \(t_{1 / 2}=-\left[\ln \left(\frac{1}{2}\right)\right] \times\left(\frac{1}{k}\right)=\frac{\ln 2}{k}=\frac{0.693}{k}\)
It is interesting to note that the half-life of a first-order reaction does not depend on the initial concentration of the reactant. Natural and artificial radioactive decay takes place through first-order reactions.
The integrated rate law for a zero-order reaction is given by
[A] = -kt +[A]0.
Substituting \([\mathrm{A}] \text { by } \frac{1}{2}[\mathrm{~A}]_0 \text { and } t \text { by } t_{1 / 2} \text {, we get }\)
⇒ \(\frac{1}{2}[\mathrm{~A}]_0=-k t_{1 / 2}+[\mathrm{A}]_0\)
or, \(+k t_{1 / 2}=[\mathrm{A}]_0-\frac{1}{2}[\mathrm{~A}]_0\)
or, \(k=\frac{1}{2} \frac{[\mathrm{A}]_0}{t_{1 / 2}}\)
or, \(t_{1 / 2}=\frac{[\mathrm{A}]_0}{2 k}\)
where [A]0 is the initial concentration of the reactant and k, is the rate constant.
The half-life of a zero-order reaction is directly proportional to the initial concentration of the reactant.
Example 1. The following data were obtained for the decomposition of N2O2, which is a first-order reaction
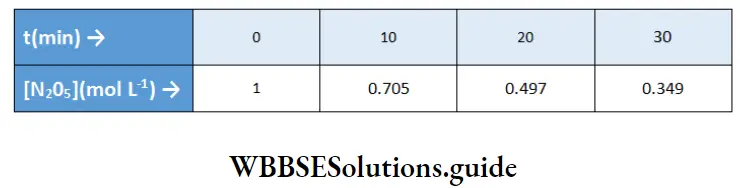
Determine the value of the specific rate constant and the half-life of the reaction.
Solution:
For a first-order reaction,
⇒ \(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=-k t\)
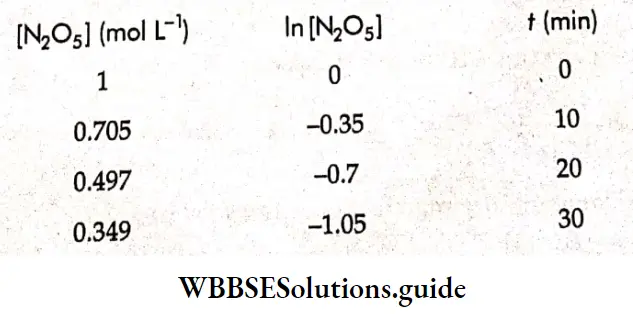
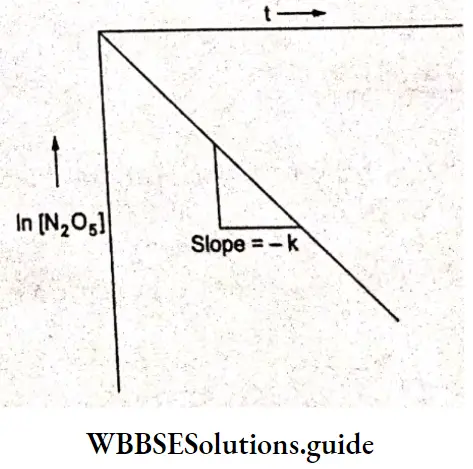
If a graph is plotted between t and ln[N205], we obtain a straight line with slope = -k.
⇒ \(\text { Slope }=\frac{Y_2-Y_1}{X_2-X_1}=\frac{-0.35}{10}=-0.035=-k\)
⇒ k =0.035 min-1.
⇒ \(\text { Half-life, } t_{1 / 2}=\frac{0.693}{k}=19.8 \mathrm{~min}\)
Example 2. The half-life for the reaction (first order)
⇒ \(\mathrm{N}_2 \mathrm{O}_5 \rightarrow 2 \mathrm{NO}_2+\frac{1}{2} \mathrm{O}_2\) is 2.4 hours at 30°C.
- Starting with 100 g of N2O2, how many grams will remain after a period of 9.6 hours?
- How much time should be required to reduce 5x 105 molecules o/N2O2 to 10 molecules?
Solution:
Half-life means the time taken for half of the reactant to disappear. If we start with 100 g, after one half-life 50 g will remain. Now of this 50 g, half the amount = 25 g will remain after another half-life, and so on.
1. After 2.4 hours,\(\frac{1}{2}\) x 100 =50 g will remain.
After 4.8 hours, \(\frac{1}{2}\) (50) = 25 g will remain.
After 7.2 hours, \(\frac{1}{2}\)(25) =12.5 g will remain.
After 9.6 hours, \(\frac{1}{2}(12.5)=\frac{1}{2} \times \frac{1}{2} \times \frac{1}{2} \times \frac{1}{2} \times 100=6.25 g\) will remain,.
We can derive a general formula here.
After 4 half-lives, \(\left(\frac{1}{2}\right)^4\) (100) g will remain.
Therefore, after n half-lives, \(\left(\frac{1}{2}\right)^n\left(C_0\right)\) of the substance will remain, where CO is the initial amount present.
2. \(t_{1 / 2}=2.4 \text { hours }\)
For a first-order reaction,
⇒ \(k=\frac{0.693}{t_{1 / 2}}=\frac{0.693}{2.4}=0.288 \text { hour }^{-1}=\frac{0.288}{60 \times 60} \mathrm{~s}^{-1}\)
⇒ \(8.02 \times 10^{-5} \mathrm{~s}^{-1}\)
⇒ \(\ln \frac{[\mathrm{A}]}{[\mathrm{A}]_0}=-k t\)
⇒ \(2.303 \log \left(\frac{10^5}{5 \times 10^5}\right)=-8.02 \times 10^{-5} \times t\)
⇒ \(t=\frac{2.303 \log \left(\frac{1}{5}\right)}{-8.02 \times 10^{-5}}=\frac{-1.6097}{-8.02 \times 10^{-5}}=20071.42 \mathrm{~s}\)
⇒ \(\frac{20071.4}{3600} h=5.57\)
It will take 5.57 hours = 5 hours 34.2 min for reducing 5 x105 molecules to 105 molecules
Example 3. The thermal decomposition of N2O2 follows first-order reaction kinetics.
2N2O2(g) 4NO2(g)+ O2(g)
Rate= k[N2O2].
What will happen to the rate if the concentration of N2Os is doubled and halved?
Solution:
Since the rate is directly proportional to [N2O5], increasing the concentration to double its value will double the value of the rate of the reaction (as k remains constant). Similarly, decreasing the concentration of N2O2 to half of its value will result in the rate being reduced to half.
Example 4. Consider the oxidation of nitric oxide
⇒ \(2 \mathrm{NO}(\mathrm{g})+\mathrm{O}_2(\mathrm{~g}) \rightarrow 2 \mathrm{NO}_2(\mathrm{~g})\)
The rate law is given as
rate= \(\text { rate }=[\mathrm{NO}]^2\left[\mathrm{O}_2\right]\)
How will the rate of the reaction be affected when the concentration of NO is doubled, NO is tripled and (c) O2 is doubled?
Solution:
The rate depends on the square of the concentration of NO.
On doubling the concentration, rate = k{2 x [NO]}2[O2 ] = k x 4[NO]2[O2]
The new rate will then be 4 times the earlier one. On tripling the concentration of NO, the rate will be 3 or 9 times the original rate. When the concentration of O2 is doubled, the rate will be doubled as the rate depends directly on the first power of O2
Determination of rate law
It is very important to note that a rate law is established experimentally and cannot in general be inferred from the chemical equation for the reaction. One of the methods for the determination of the rate law is called the Ostwald method in which the concentration of one of the reactants is much less than that of the others. Consider a reaction with two reactants A and B, If the reactant B is taken in excess, then even when the reaction reaches completion, the concentration of B is only marginally affected. It is a good approximation to take its concentration as a constant throughout the reaction. Even if the true rate law is
rate = k[A][B],
we say that rate =k[A] where k’ = k[B]0 where [B]0 is the initial concentration of the reactant B, which hardly changes. Since the true law has been forced into an effective first-order form, it is called a pseudo-first-order rate law and the reaction under such conditions is a pseudo-first-order reaction.
For example, consider the reaction between ozone and NO.
O3 + NO → O2 + NO2
or that between O3 and NO2.
O3+ 2NO2 → 2N3 + O2
Both these reactions are of the second order overall and of the first order with respect to each reactant.
Rate = k[O3][NO].
Rate = k[O3][NO2].
The integrated rate law for a reaction that is of the first order with respect to two reactants is complicated.
However, a simple rate law expression can be derived for such a reaction when the concentration of one of the reactants is much higher than that of the other. The concentration of O3 is generally hundreds to thousands of times greater than the concentration of NO in polluted air. The concentration of ozone remains more or less constant during the course of the reaction.
The rate law can be simplified to
rate = k[NO],
where k = k[O3]0 where [O3]0 is the initial concentration of ozone.
The above rate law looks like the rate law of a first-order reaction; it appears to obey first-order kinetics. It is hence called a pseudo-first-order reaction and the rate constant is called the pseudo-first-order rate constant.
Temperature dependence of reaction rate
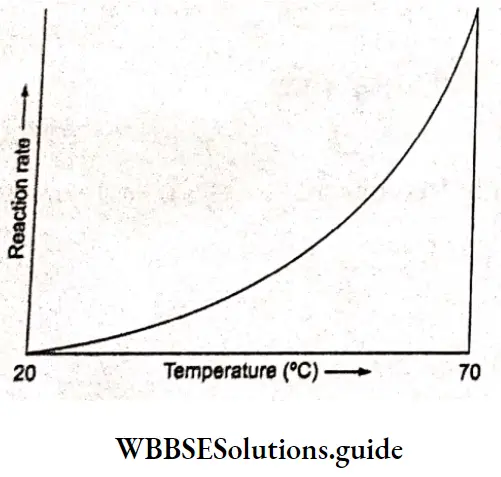
For many chemical reactions, the rate increases as the temperature is raised. You must have noticed that curd sets faster in summer than in winter. When the temperature is relatively high metabolic reactions are faster. It is said that fever is our friend because we fight infection with a fever.
The rise in the body temperature upsets the balance of mission rates in the foreign organism, thus preventing it from further invading the body. Also, an increase in the temperature of the body kills the invading organism. Reaction rates are found to increase exponentially with an increase in temperature. In general, for a 10 C rise in temperature, the reactions become two or three times faster. For example, the rate of hydrolysis of sucrose is 4.1 times more at 35’Cl than at 25cC.
Arrhenius’s equation:
In 1889 Arrhenius studied the data accumulated on reaction rates and found that the rate constant k of most chemical reactions increases exponentially as a function of temperature, T. The mathematical form of this relation is called Arrhenius’s equation, which is as follows.
⇒ \(k=A \exp \left(-\frac{E_a}{R T}\right)\)
Here, k is the rate constant, A is called the pre-exponential factor, frequency factor, or collision frequency. It is also simply called the Arrhenius constant. Ea is called the activation energy. The term exp \(\left(-\frac{E_a}{R T}\right)\) represents an activation state factor. Collectively these quantities (A and Ea) are called Arrhenius parameters.
Activation energy:
Activation energy is the minimum energy required for a chemical reaction to take place. It is the minimum kinetic energy required by reactant molecules to break bonds and form new ones (those of products).
To understand the concept of activation energy better, consider a boulder that has to be moved up a hill.
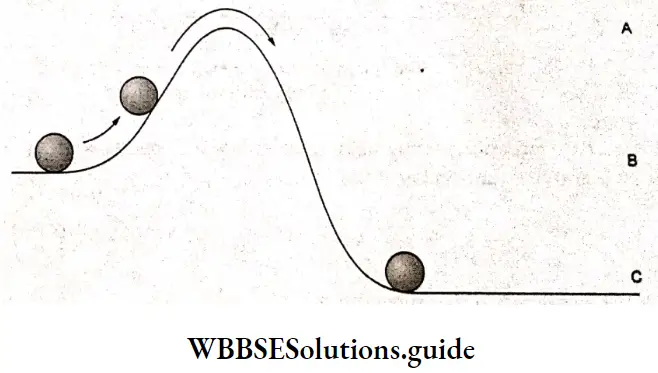
The boulder initially at position B loses some of its potential energy when it reaches position C. However, it should have sufficient kinetic energy to cross the barrier at A. As the boulder moves from position B to A, it gradually loses its kinetic energy and gains potential energy.
If the kinetic energy of the boulder is higher than its potential energy at A, it can cross and reach the other side of the hill. From here, it can simply slide down to reach C, The minimum energy required by the boulder to cross the barrier is the activation energy. It is the difference between the energy at B and that at A.
Let us now consider the following chemical reaction.
A + BC → AB + C
If the reaction occurs in a single step, the reactant molecules collide with each other, and the electron distribution about the three nuclei (A, B, and C) changes in the course of a collision such that a new bond between A and B is formed and at the same time the bond between B and C breaks.
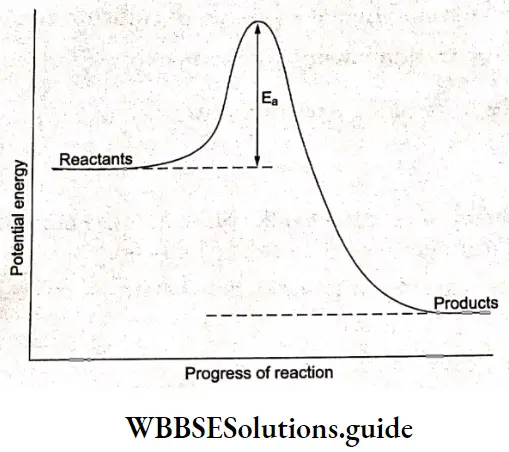
Between the reactant stage and product stage, the nuclei pass through a stage in which all three species (A, B, and C) are weakly linked together. This has a higher potential energy than both reactants and products.
The reactants must gain enough energy to overcome this energy barrier. The energy actually comes from the ‘ kinetic energy of the molecules which is converted into potential energy. The height of the potential energy barrier is called activation energy.
Taking the natural logarithm of both sides of Arrhenius’s equation, we get
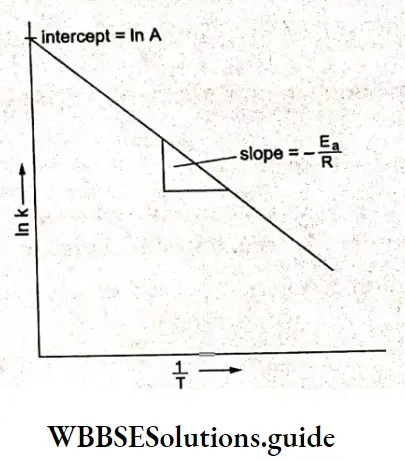
⇒ \(\ln k=\ln A-\frac{E_a}{R T}=\ln A-\left(\frac{E_a}{R}\right)\left(\frac{1}{T}\right)\)
A graph plotted between In k (In of rate constant) and \(\frac{1}{T}\) where T is the absolute temperature at which k is measured, is a straight line with a slope equal to \(-\frac{E_a}{R}\). The intercept is In A, from which A can be calculated.
Activation energy may also be calculated if the rates are available at only two temperatures. At temperature T1, the above equation becomes
⇒ \(\ln k_1=\ln A-\left(\frac{E_a}{R}\right) \cdot \frac{1}{T_1}\)
At temperature T2, it becomes
⇒ \(\ln k_2=\ln A-\left(\frac{E_a}{R}\right) \cdot \frac{1}{T_2}\)
Subtracting the first equation from the second,
⇒ \(\ln k_2-\ln k_1=-\frac{E_a}{R}\left(\frac{1}{T_2}-\frac{1}{T_1}\right) \text { or } \ln \left(\frac{k_2}{k_1}\right)=-\frac{E_a}{R}\left(\frac{1}{T_2}-\frac{1}{T_1}\right)\)
This equation can be used to calculate Ea from the rate constants and k2 at temperatures Ta and T2.
Example 1. The rate constant first-order reaction becomes five times the original rate constant when the temperature rises from 350 K to 400 K. Calculate the activation energy for the reaction.
Solution:
Given
The rate constant first-order reaction becomes five times the original rate constant when the temperature rises from 350 K to 400 K.
⇒ \(\ln k=\ln A-\frac{E_a}{R T}\) (Arrhenius’s equation)
At T = 350 K, let the rate constant = k.
∴ at T = 400 K, the rate constant = 5k.
Substituting the values at 350 K and 400 K,
⇒ \(\ln k=\ln A-\frac{E_a}{8.314 \times 350}\)
and In (5k) = \(\ln A-\frac{E_a}{8.314 \times 400}\)
Subtracting Equation (1) from Equation (2), we have
⇒ \(\ln (5 k)-\ln k=-\frac{E_a}{8.314 \times 400}+\frac{E_a}{8.314 \times 350}\)
⇒ \(\ln \left(\frac{5 k}{k}\right)=\frac{E_a}{8.314}\left(\frac{-1}{400}+\frac{1}{350}\right)\)
⇒ \(\ln 5=\frac{E_a}{8.314}\left(-2.5 \times 10^{-3}+2.857 \times 10^{-3}\right)\)
⇒ \(\frac{1.609 \times 8.314}{3.57 \times 10^{-4}}=E_a\)
⇒ \(E_a=37.47 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Example 2. The rate constants for the decomposition of HI at different temperatures are as follows.
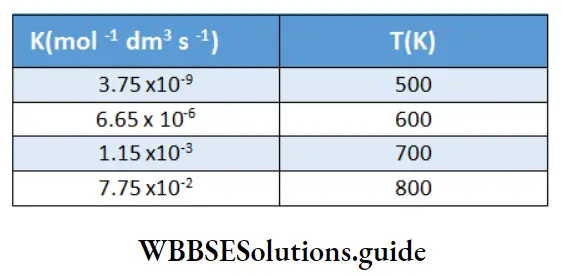
Find the value of the activation energy for this reaction.
Solution:
A plot of In k vs \(\frac{1}{T} \text { gives }\left(-\frac{E_a}{R}\right)\) as the slope.

⇒ \(\text { Slope }=-22727.2=-\frac{E_a}{R}\)
⇒ \(E_a=R \times 22727.2=8.314 \times 22727.2=188954.5 \mathrm{~J} \mathrm{~mol}^{-1}\)
or, \(E_a=188.9 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
The activation energy for the decomposition of HI is 188.9 kJ mol-1.
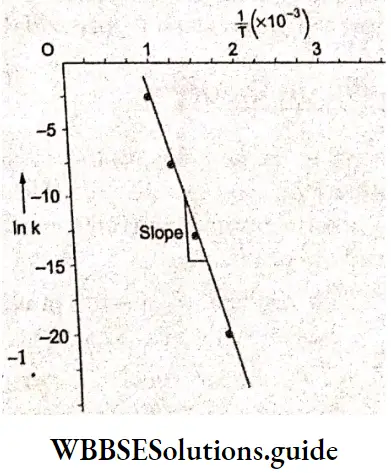
Effect of catalyst
Yet another very important factor determining the rate of a reaction is the presence of a catalyst. A catalyst is a substance that increases the rate of a reaction without itself undergoing any permanent chemical change. An example is manganese dioxide, which speeds up the thermal decomposition of potassium chlorate.
⇒ \(2 \mathrm{KClO}_3(\mathrm{~s}) \stackrel{\mathrm{MnO}_2 \text {, heat }}{\longrightarrow} 2 \mathrm{KCl}(\mathrm{s})+3 \mathrm{O}_2(\mathrm{~g})\)
In the absence of a catalyst, KClO3 decomposes very slowly, even when heated, but when a small amount of MnO2
is mixed with the KClO3 before heating, the rapid evolution of oxygen takes place. The MnO2 can be recovered unchanged after the reaction is complete.
Most industrial processes, such as the manufacture of ammonia, sulphuric add, nitric acid, and polymers, involve the use of catalysts. Biological catalysts or enzymes affect the rates of reactions taking place during metabolic activities.
How does a catalyst work? It increases the rate of a reaction by lowering the activation energy. The potential energy barrier between the reactants and products is reduced, thus facilitating a faster reaction (Figure 4.13). A catalyst participates in the chemical reaction by forming an intermediate complex with the reactants, which finally breaks down to form the products, and the catalyst is obtained back.
Temperature and catalyst effects on reaction rate
The concentration of the catalyst does not appear in the rate law of any reaction because while it is consumed in one step, it is regenerated in another step. Let us consider the decomposition of H2O2
⇒ \(2 \mathrm{H}_2 \mathrm{O}_2(\mathrm{aq}) \rightarrow 2 \mathrm{H}_2 \mathrm{O}(\mathrm{l})+\mathrm{O}_2(\mathrm{~g})\)
This reaction has a high activation energy of 76 kJ mol-1 at room temperature and hence this decomposition is very slow. In the presence of the iodide ion, the reaction is appreciably faster because it proceeds by a different, lower-energy, pathway.
\(\begin{gathered}\mathrm{H}_2 \mathrm{O}_2(\mathrm{aq})+\mathrm{I}^{-}(\mathrm{aq}) \rightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l})+\mathrm{IO}^{-}(\mathrm{aq}) \\
\mathrm{H}_2 \mathrm{O}_2(\mathrm{aq})+\mathrm{IO}^{-}(\mathrm{aq}) \rightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l})+\mathrm{O}_2(\mathrm{~g})+\mathrm{I}^{-}(\mathrm{aq}) \\
\hline 2 \mathrm{H}_2 \mathrm{O}_2(\mathrm{aq}) \rightarrow 2 \mathrm{H}_2 \mathrm{O}(\mathrm{l})+\mathrm{O}_2(\mathrm{~g})
\end{gathered}\)
The H2O2 first oxidizes the catalyst I– to the hypoiodite ion (IO–) and then reduces the intermediate IO– back to I-1. The activation energy is, thus, lowered by 19 kJ mol-1.
Collision Theory and Transition State Theory
Two theories have been put forward which explain the mechanistic and energetic aspects of chemical reactions. These are the collision theory and the transition state theory.
Collision theory:
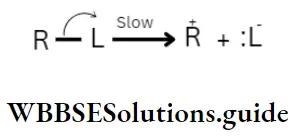
According to the collision theory, an atom, ion or molecule can undergo a reaction only when it collides with another atom, ion or molecule. The reacting atoms, ions or molecules are assumed to be hard spheres. However, only a small number of collisions result in a chemical reaction. The following conditions must be fulfilled for a collision to be effective.
1. The transfer or sharing of electrons between the colliding species must give a structure that is capable of existence. In other words, stable bonds or new stable chemical species should be formed.
2. The collision must take place with sufficient energy—the outer electronic shells of the atoms should penetrate each other to some extent so that the bonding electrons can be rearranged.
3. The orientation of the molecules when they collide must that the atoms directly involved in the sharing or transfer of electrons come into contact with each other
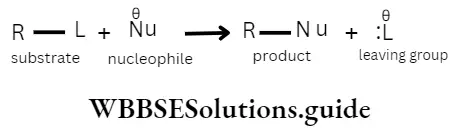
Now, let us consider these three aspects in a rate equation of a bimolecular elementary reaction(see section or reaction mechanisms) A + B → P.
The rate of the reaction can be expressed as
Rate = f x P x Z [A][B]
where P and Z represent the three aspects of collision theory, [A] and [B] are the reactant concentrations. The rate constant k then takes the form.
k = f x P x Z
To understand what ‘f’ denotes, let us see the effect of temperature on the above reacting system Changing the temperature of a reacting system has two effects.
On raising the temperature, molecules tend to move faster and hence there are a larger number of collisions but calculations show that only a small percentage of the rate increase with temperature can be accounted for by the larger number of collisions. The more important factor is the kinetic energy of the molecules and it is found that the average kinetic energy of molecules increases with an increase in temperature.
In other words, the activation energy required for the reaction to take place is provided by the collisions of the reactant molecules with each other or with the walls of the reaction vessel. Only a few fast-moving molecules will have enough energy to react if the activation energy is larger than the average kinetic energy of the molecules. The kinetic energy is converted to the potential energy of the molecule.
If the activation energy is smaller than the kinetic energy, then the fraction of molecules having the required kinetic energy will be large and most collisions will result in the reaction. As a result, the reaction will be fast.
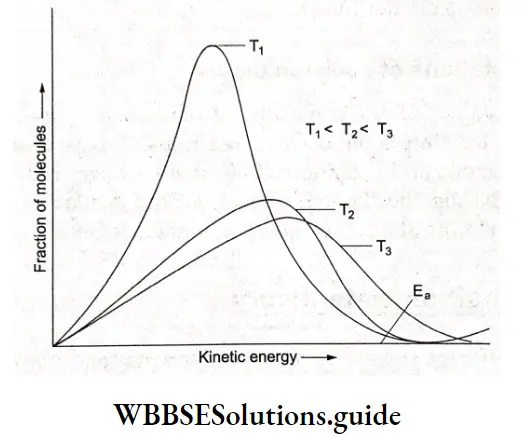
The fraction of molecules (f) that possesses kinetic energy equal to or greater than Ea is equal to \(e^{-E_f / k T}\) as in Arrhenius’s equation. As the temperature increases, the curve broadens resulting in an increase in the fraction of molecules having an energy Ea, As T increases, \(\frac{1}{T}\) decreases, and f increases exponentially, Z in Equation 4.7 is called the collision frequency and is defined as the number of collisions per second per unit volume of the reaction mixture.
For collisions between reactant molecules A and B, Z is represented as Though a large fraction of molecules may possess kinetic energy greater than the activation energy, all collisions between such molecules may not lead to products. This is the third aspect of collision theory.
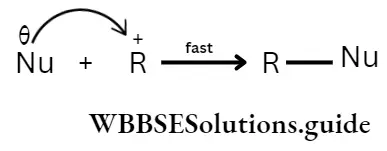
The fraction of collisions that lead to products is restricted by the requirement of proper orientation of the colliding molecules. This is represented by the probability or steric factor P, which denotes the fraction of collisions in which the reacting molecules have the right orientation to form the product.
For example, in the reaction, between HCl and NH3 to form NH4Cl, the N end of NH3 must hit the H end of HCl. Such a collision which results in the formation of a product is called an effective collision. Collision of the N of NH3 with Cl of HCl does not result in Hr formation of NH4Cl and hence it is termed as ineffective collision. The following shows an effective and an ineffective collision for the reaction between HCl and NH-.,

The value of P is usually between 0 and 1. The rate constant can thus be written as
k = fPZAB = PZAB e-2Ea/RT
It is interesting to note that tins resemble Arrhenius’s equation (Equation 4.6) and by a comparison of those two equations, we got
A = PZAB
where A is the Arrhenius factor.
Arrhenius’s equation reflects two aspects of the collision theory of reaction rates. The frequency factor A indicates the number of collisions that lead to a proper orientation of the reactant species to enable the formation of products, The exponential term \(e^{\left(-E_a / RT\right)}\) the fraction of collisions in which the energy of the reacting spades is greater than Ea
Limitation of collision theory:
The values of rate constants calculated using the collision theory are in agreement with the experimental values only for simple bimolecular reactions. This is because the molecules are considered to be hard spheres and the vibrations and rotations within them are ignored. There is no way of determining P. It can only be obtained by comparing the theoretically calculated A (Arrhenius factor) and the experimentally observed value. You will study more about this theory in higher classes,
Transition state theory
A different theory as to how reactions take place pictures the formation of a transition slate by the two (or more) reacting molecules instead of their collision. The molecules undergoing reactions are considered to be approaching each other and under each other’s influence, a transition state is formed.
The tire transition state or the activated complex is a combination of reacting molecules, intermediate between reactants and products. Some bonds begin to break while new ones begin to form.
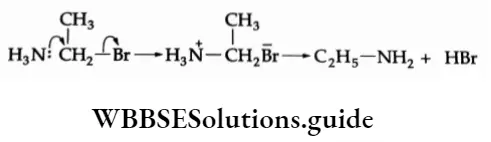
For example, in the reaction between ammonia and methyl bromide, the N-C bond begins to form in the transition state, the C-Br bond begins to weaken, and partial charges develop on tire nitrogen and bromine atoms.
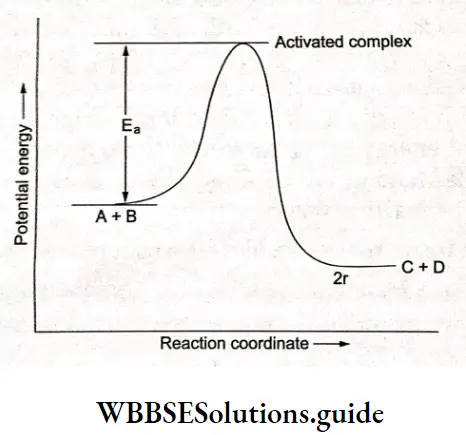
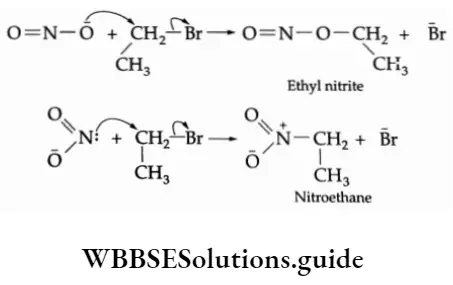
⇒ \(\mathrm{NH}_3+\mathrm{CH}_3 \mathrm{Br} \longrightarrow\left[\mathrm{CH}_3 \mathrm{NH}_3\right]^{+}+\mathrm{Br}^{-}\)
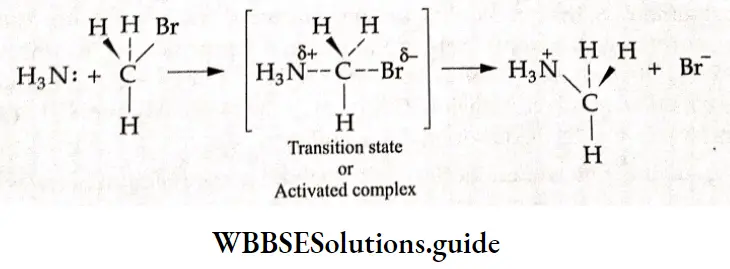
An activated complex cannot ordinarily be isolated. It breaks down to give either reactants or products depending on the conditions of the reaction.
The energy of the transition state is higher than that of the two reactants by an amount of Ea —the activation energy. For the reaction to take place, the sum of tire kinetic energies of A and B must be will least equal to or greater than E„.
After reaching the transition state, the molecules start losing energy and ultimately reach a state of energy even lower than that of the initial state. The energy thus lost is transferred to other molecules and they reach the transition state; the reaction proceeds in this the size of the activation energy barrier depends on the direction from which it is approached.
If the reaction is exothermic in the forward direction, Ea is smaller in the forward direction than in the reverse direction. It is easier to cross the smaller energy barrier in the case of the exothermic reaction than in the reverse endothermic reaction.
Difference between average rate and instantaneous rate of reaction
At the same temperature, some reactions have a high activation energy and some have a low activation energy. If we compare two reactions at the same temperature, the one with a lower activation energy will be fast and have a high rate constant whereas the one which has a higher activation energy will have lower k and lower rate.
Increasing the temperature increases the value of \(e^{\left(-E_a / R T\right)}\) and hence gives a larger k. A rise in temperature also increases A. Any change in the conditions of the reaction that increases the number of collisions with a favorable orientation of the molecules results in a high value of k.
Reaction mechanisms
Most chemical reactions occur in a series of elementary reaction steps, rather than in a single step.
The process or pathway by which a reaction occurs actually is called the reaction mechanism or reaction path.
A balanced chemical equation does not tell us anything about the actual pathway (steps in the reaction); it just tells us what molecules and how many of them react to form what kind of products. The mechanism of a reaction can be deduced from a knowledge of the rate law of that reaction.
As already discussed, the rate law and order of a reaction have to be experimentally determined, and once determined, a mechanism for the reaction can be suggested. Here, we shall look at the mechanisms of some simple reactions.
First, consider the example of the decomposition of ozone. The chemical equation for the overall reaction is
2O3 → 3O2
The reaction, however, appears to follow a mechanism involving two steps.
O3 → O2 + O
O+O3 → 2O2
Atomic oxygen is produced in the first step and is consumed in the second step. Hence it is not shown in the overall reaction. Such species which are produced in one step and consumed in another are called intermediates.
Most reactions proceed in several steps involving one or more reaction intermediates. These intermediates may be short-lived or may persist. The rate of such a multistep reaction is determined by the slowest step.
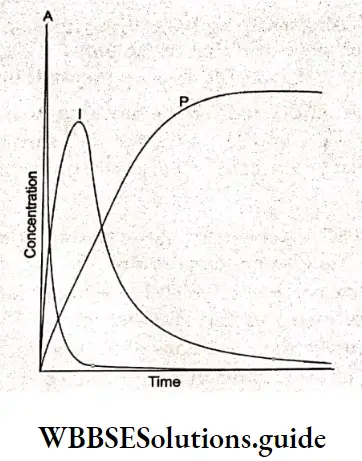
Now consider a reaction involving two consecutive steps of the first order. If in a chemical reaction, a reactant produces an intermediate (I) which decays to form the product (P), we can say that the reaction takes place in two steps.
A → I Rate of formation of I = k1[A]
I → P Rate of formation of P = k2[I]
Shows that the concentration of the intermediate (I) grows and reaches a maximum initially.
Meanwhile, the concentration of the product also rises. After the concentration of the intermediate reaches a maximum, it decays to zero and that of the product reaches its final value.
An example of such a reaction is the radioactive decay of uranium. The half-lives of uranium and neptunium are given.
⇒ \({ }^{239} \mathrm{U} \stackrel{23.5 \mathrm{~min}}{\longrightarrow}{ }^{239} \mathrm{~Np} \stackrel{2.35 \text { days }}{\longrightarrow}{ }^{239} \mathrm{Pu}\)
An intermediate may also be formed when we have two reactants; an example of such a reaction is the enzyme-catalyzed reaction in which a substrate S is converted to products. The proposed mechanism is
⇒ \(\mathrm{E}+\mathrm{S} \rightleftharpoons \mathrm{ES} \rightarrow \text { products }+\mathrm{E}\)
where E is the enzyme and ES is the intermediate, a state in which the enzyme is bound to the substrate.
The rate-determining step:
Reactions that take place in more than one step are called complex reactions. Each complex reaction is generally a combination of a sequence of elementary reactions, which we refer to as steps. The overall balanced equation for a reaction is the sum of its elementary steps.
Elementary reactions individually may involve one or more molecules. Those involving a single molecule are called unimolecular while those that involve two molecules are called bimolecular. Similarly, there may be tennolecular reactions (involving three molecules), and so on.
The molecularity of an elementary reaction refers to the number of reacting particles, viz., atoms, molecules, or ions, in that particular step. The order and molecularity of a reaction are two different aspects.
While the order is determined experimentally and may or may not be the same as the stoichiometry of the molecule under consideration, the molecularity of the reaction can be judged from the stoichiometry of a particular step of a reaction.
The order of a reaction is applicable to an elementary as well as a complex reaction while molecularity is applicable only to an elementary reaction. The order may be zero or fractional but molecularity can only be a positive integer. The order is determined by the slowest step in a complex reaction, i.e., the rate-determining step.
Generally, the molecularity of the rate-determining step is the same as the order of the overall reaction. To understand this better, let us consider the mechanism for the oxidation of the iodide ion by H2O2 in an acid medium and find the rate equation. For the reaction
⇒ \(\mathrm{H}_2 \mathrm{O}_2+2 \mathrm{H}^{+}+2 \mathrm{I}^{-} \rightarrow \mathrm{I}_2+2 \mathrm{H}_2 \mathrm{O}\)
the suggested mechanism is
⇒ \(\mathrm{H}_2 \mathrm{O}_2+\mathrm{I}^{-} \rightarrow \mathrm{OH}^{-}+\mathrm{HOI}\) (slow)
⇒ \(\mathrm{H}^{+}+\mathrm{OH}^{-} \rightarrow \mathrm{H}_2 \mathrm{O}\) (fast)
⇒ \(\mathrm{HOI}+\mathrm{H}^{+}+\mathrm{I}^{-} \rightarrow \mathrm{I}_2+\mathrm{H}_2 \mathrm{O}\) (fast)
Since the slowest step determines the rate, the rate equation for the reaction is
⇒ \(\text { rate }=-\frac{d\left[\mathrm{H}_2 \mathrm{O}_2\right]}{d t}=k\left[\mathrm{H}_2 \mathrm{O}_2\right]\left[\mathrm{I}^{-}\right]\)
Let us consider one more example and see how the rate law is In agreement with the rate-determining step.
Consider the decomposition of NO2.
2NO2(g) → 2NO(g) + O2(g)
This reaction is of the second order with respect to NO2. The two steps in which this reaction occurs are
2NO2 → NO+NO3
NO3 → NO+O2
In the first step, a collision between a pair of NO2 molecules produces a short-lived activated complex in which the two molecules share an oxygen atom. Such activated complexes have a lifetime of ~ 10 -15 s and fall apart forming products or reactants.
To form products, the shared oxygen is transferred from one NO2 molecule to another forming a molecule of NO and a molecule of NO2. The NOa thus formed decomposes in the second step again by the formation of another activated complex.
NO2 is called the intermediate in this mechanism as it is produced in one step and consumed in the next. Intermediates are neither considered as reactants nor as products and hence do not appear in the equation describing the reaction.
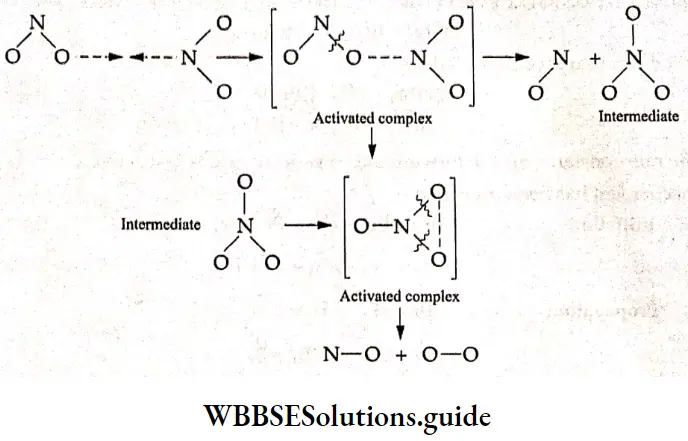
The lines between the atoms just show that a bond exists between the two—the nature of the bond is not shown.
The observed rate law which indicates an order of 2 with respect to NO2 can be predicted for step 1 but not for step 2. This means that the rate of the overall reaction is controlled by step 1 rather than step 2. The first step is the controlling step which is also the slower of the two in this case and is called the rate-determining step.
The rate for the first step is
⇒ \(\text { rate }=-\frac{\Delta\left[\mathrm{NO}_2\right]}{\Delta t}=k_1\left[\mathrm{NO}_2\right]^2\)
If the rate constant k1 is much less than that for step 2, k2, then
⇒ \(\text { rate }=-\frac{\Delta\left[\mathrm{NU}_3\right]}{\Delta t}=k_2\left[\mathrm{NO}_3\right]\)
In fact, it has been found that
k1 << k2.
In an unimolecular reaction, a single molecule shakes itself apart or its atoms into a new arrangement as in the isomerization of cyclopropane to propene.
⇒ \(\underset{\text { Cyclopropane }}{\Delta} \rightarrow \mathrm{CH}_2=\underset{\text { Propene }}{\mathrm{CH}}-\mathrm{CH}_3\)
An unimolecular elementary reaction is of the first order. For example in a reaction A → B,
⇒ \(\frac{d[\mathrm{~A}]}{d t}=-k[\mathrm{~A}]\)
In a bimolecular elementary reaction, two molecules collide with each other and then react. Such a reaction is of second order; the rate is proportional to the concentrations of the two reactants.
For a reaction of the type A + B → product
⇒ \(\frac{d[\mathrm{~A}]}{d t}=-k[\mathrm{~A}][\mathrm{B}]\)
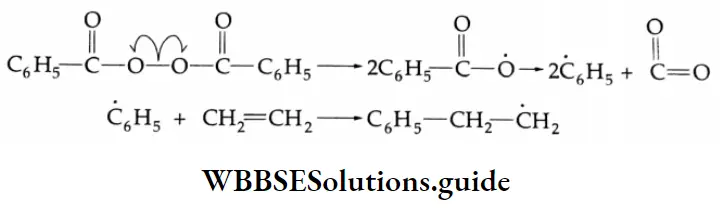
Chain reactions:
A chain reaction is a complex reaction comprising a sequence of reactions in which the intermediate formed in the first step generates a reactive intermediate in the subsequent step, and so on. The intermediates responsible for the propagation of a chain reaction are called chain carriers. They may be radicals ions, or neutrons in nuclear fission reactions.
The steps in a chain reaction are classified as the initiation step, propagation step, and termination step. There can be more than one propagation or termination step possible for a chain reaction. Many gas-phase reactions and liquid-phase polymerization reactions are chain reactions. The formation of HBr from H2 and Br2 is a chain reaction.
⇒ \(\mathrm{H}_2(\mathrm{~g})+\mathrm{Br}_2(\mathrm{~g}) \rightarrow 2 \mathrm{HBr}(\mathrm{g})\)
Experimental methods to determine reaction rate
The rate law for the reaction is complicated.
⇒ \(\frac{d[\mathrm{HBr}]}{d t}=\frac{k\left[\mathrm{H}_2\right]\left[\mathrm{Br}_2\right]^{3 / 2}}{\left[\mathrm{Br}_2\right]+k^{\prime}[\mathrm{HBr}]}\)
where k and K are the rate constants for the forward and reverse reactions respectively.
The following mechanism has been proposed.
Initiation \(\mathrm{Br}_2+\mathrm{Br}_2 \rightarrow \mathrm{Br}+\mathrm{Br}+\mathrm{Br}_2\)
or \(\mathrm{Br}_2+\mathrm{H}_2 \rightarrow \dot{\mathrm{Br}}+\dot{\mathrm{Br}}+\mathrm{H}_2\)
Propagation \(\dot{\mathrm{Br}}+\mathrm{H}_2 \rightarrow \mathrm{HBr}+\dot{\mathrm{H}}\)
or \(\dot{\mathrm{H}}+\mathrm{Br}_2 \rightarrow \mathrm{HBr}+\dot{\mathrm{Br}}\)
Retardation \(\dot{\mathrm{H}}+\mathrm{HBr} \rightarrow \mathrm{H}_2+\dot{\mathrm{Br}}\)
Termination \(\dot{\mathrm{Br}}+\dot{\mathrm{Br}}+\mathrm{H}_2 \rightarrow \mathrm{Br}_2+\mathrm{H}_2^*\)
\(\dot{\mathrm{Br}}+\dot{\mathrm{Br}}+\mathrm{Br}_2 \rightarrow \mathrm{Br}_2+\mathrm{Br}_2^*\)A dot indicates a free radical and a star indicates an activated molecule.
As you already know, free radicals are highly reactive. The chain reaction continues until the termination step occurs. In this step, either Br2 or H2 removes the excess energy of recombination and exists as an activated (high-energy) molecule.



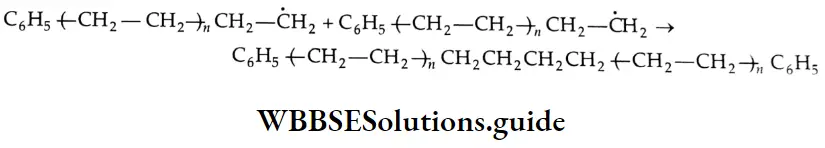



















![Coordination Compounds and Organometallics The cis and trans isomers of the square-planar [Ma2b2] type and The cis and trans isomers](https://wbbsesolutions.guide/wp-content/uploads/2023/10/Coordination-Compounds-and-Organometallics-The-cis-and-trans-isomers-of-the-square-planar-Ma2b2-type-and-The-cis-and-trans-isomers.png)