
Crystal-Field Theory (CFT)
Crystal-Field Theory (CFT):
This theory was proposed by Bethe and Van Vleck. In this theory, the attraction between the central atom and ligands is assumed to be purely electrostatic. The theory is very useful in explaining the magnetic behaviour and electronic spectra of transition metal complexes.
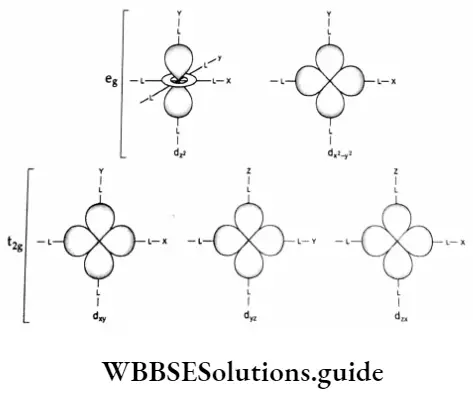
Before discussing the CFT it is worthwhile to study the shapes of d orbitals because the crystal field treatment of coordination compounds is based on the spatial relationships of the d orbital to the surrounding ligands in an asymmetric field.
In CFT it is assumed that ligands are point charges if they are anions or point dipoles if they are neutral molecules. The other assumption is that there is no interaction between metal and ligand orbitals. In other words, there is no allowance for covalence in metal-ligand bonds.
Crystal field theory class 12 chemistry notes
The crystal-field theory may be applied to coordination complexes to answer the question as to how the energies of a set of d orbitals split when a set of ligands is placed around a central metal ion.
The d orbitals of an isolated gaseous metal atom or ion have the same energy, i.e., they are degenerate. If a spherically symmetrical field of negative charge surrounds the metal ion, the d orbitals remain degenerate.
However, in the presence of an asymmetric field (as in complexes), this degeneracy disappears, as the d orbitals, by virtue of the difference in their shapes, are not affected equally. This phenomenon is known as crystal-field splitting. Here we shall discuss the octahedral and tetrahedral complexes.
Octahedral complexes
The octahedral complex is the simplest case to consider. Assume that the metal atom is at the centre surrounded by six ligands at the vertices of an octahedron. For convenience, the arrangement is defined relative to a set of cartesian axes x, y and 2. The metal atom is at the origin and the ligands are positioned symmetrically along the cartesian axes.
Compare the shapes of the d orbitals with the octahedral arrangement of ligands. As you can see the lobes of the orbitals dx2-y2 and dz2 point along the x, y and z axes while those of the orbitals,dxy,dxz and dyz point between the axes.
This shows that the orbitals dx2-y2 and dz2 on the one hand and the orbitals, dxy,dxz and dyz on the other hand have equivalent relationships to the set of ligands. Therefore, we may say that five d orbitals form two sets each containing two and three orbitals respectively.
Then it should be obvious that the electron present in any one of the orbitals of a set will be repelled to the same extent by the ligands. Thus, the orbitals within a set are equivalent in energy or degenerate (remember this is not the case when a spherically symmetrical field surrounds the metal atom).

As the ligands approach the metal atom, the orbitals lying along the axes (dz2 and dx2-y2 ) get more strongly repelled than those which have lobes directed between the axes (dxy,dxz and dyz). Thus, one set of orbitals gets raised in energy and the other is lowered relative to the average energy in the spherical crystal field.
Therefore one conclusion of the crystal-field theory is that the spatial relationships of the d orbitals to the surrounding ligands cause the five d orbitals to split into two sets.
Definition and explanation of crystal field theory
The orbitals dxy,dyz and dzx with lower energy are known as the t2g orbitals. The orbitals dz2 and dx2-y2 are known as e8 orbitals.
In an octahedral field, the electrons present in eg orbitals experience greater repulsion than the electrons in t2g orbitals.
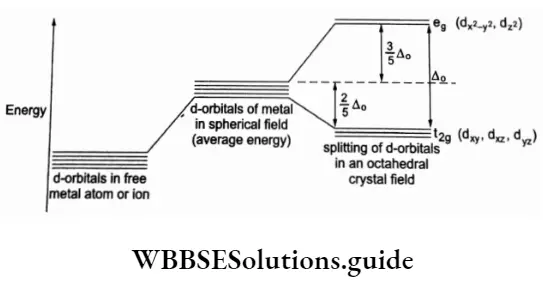
The difference in energy between the two sets of d orbitals is denoted by Δ0 (the symbol o in the subscript stands for octahedral) and is called crystal field stabilisation energy (CFSE). On splitting, the energy of two eg orbitals increases by (3/5) A0 and that of the three t2g orbitals decreases by (2/5) Δ0.
When a metal has only d electron (d ion), for example, [Ti(H2O)6]3+, then the electron is present in one of the lower t2g orbitals. In d2 and d3 entities, the three t2g orbitals are filled singly by Hund’s rule.
Crystal field theory with examples and diagrams
For a d4 ion, there are two possibilities -the fourth electron may enter an eg orbital of higher energy (t2g3eg1) or may pair an electron in a t2g orbital (t2g4 ).
The exact configuration adopted is governed by the relative magnitudes of Δ0 and P. (P is the energy needed to cause the pairing of an electron in an orbital.)

If Δ0 < P, then the fourth electron goes to the eg orbital as the energy needed for pairing is more. This is called a weak-field, high-spin situation. If A Δ0> P then pairing occurs in t2g orbitals. This is called a strong-field, low-spin situation. The strong-field situation is generally more stable than the weak-field situation.

The configuration of coordination entities with four to seven d electrons (the strong-field situation is more stable than the weak-field situation)
Tetrahedral Complexes
In tetrahedral complexes the direction of approach of ligands is different; the t2g orbitals are closer to the ligands than the eg orbitals. Thus the crystal field splitting in tetrahedral complexes is the opposite to that in octahedral complexes.
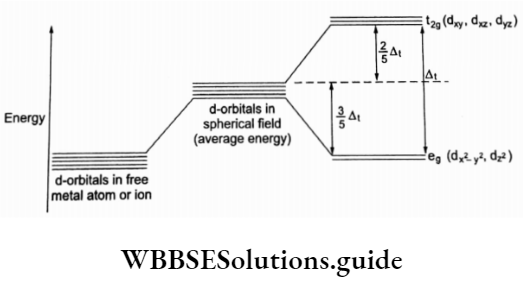
The magnitude of crystal field splitting Δt is less than Δ0. \(\left(\Delta_t=\frac{4}{9} \Delta_0\right)\) (This number \(\left(\frac{4}{9}\right)\) has been obtained from spectroscopy). Δ0 is the energy difference between d orbitals in the octahedral field whereas At is the energy difference in the tetrahedral field.
Crystal field splitting energy and its significance
The magnitude of crystal field splitting depends on the
- Nature of the ligands
- Charge on the metal ion (generally A increases with charge)
- Position of the metal in the periodic table—whether it is in the first, second or third row of transition elements. The general trend for Δ is 3d < 4d < 5d. Thus, heavier transition metals generally form low-spin complexes.
Ligands causing small crystal-field splitting are called weak-field ligands while those causing large crystal-field splitting are called strong-field ligands.
The common ligands can be arranged in ascending order of A. This order is constant for different metals and is called the spectrochemical series.
weak-field I- < Br–< S2– <cI– < NO3– < F–< OH- <C2O42-< H2O< EDTA < NH3 and pyridine < en < NO2– < CN–< CO strong-field
Magnetic Properties Of Coordination Compounds
Coordination compounds may be paramagnetic or diamagnetic. Paramagnetism arises due to the presence of unpaired electrons and if no unpaired electrons are present, the compound is diamagnetic.
The magnetic moment of a coordination compound, which is related to the number of impaired electrons, is an experimentally determined value and is related to the number of unpaired electrons.
Crystal field splitting in octahedral and tetrahedral complexes
A study of the magnetic moment values of complexes of two metals of the 3d series reveals some interesting facts. For metal ions containing up to three electrons in the d orbitals, there is a direct relation between the magnetic moment values and the number of d electrons.
For example, in Ti3+ (d1 ), V3+ (d2 ) and Cr3+ (d3 ), the magnetic moment of the coordination compound is equal to that of the corresponding free ion. For these metal ions, two vacant 3d orbitals are available to hybridise with one 4s and three 4p orbitals to give six hybrid orbitals needed for octahedral geometry.
However, when the metal ion has more than three d electrons, the required number of d orbitals needed for octahedral hybridisation (d2 sp3 ) is not available. A vacant pair of d orbitals in such cases may result from the redistribution of electrons in d orbitals.
Thus, in a d4 system (Cr2+, Mn3+) one of the d electrons pairs up leaving two impaired electrons, while in d5(Mn2+, Fe3+) and d6 (Fe2+, Co3+) systems two and three electrons pair up leaving one and zero unpaired electrons respectively. Hence, in such cases, the magnetic moment of the coordination compound does not tally with that of the free metal ion.
In reality, all coordination compounds involving d4, d5 and d6 metal ions do not show similar magnetic properties. [Mn(CN)6]3– has a magnetic moment corresponding to two impaired electrons, while [MnF6]3- has a magnetic moment corresponding to four unpaired electrons.
Magnetic studies reveal that [Fe(H2O)6]3+ has five unpaired electrons, while [Fe(CN)6]4- has one unpaired electron. [Co(NH3)6]3+ is diamagnetic while [CoF6]3- has four unpaired electrons. Thus, the magnetic behaviour changes with the nature of the ligand.
Octahedral crystal field splitting diagram and explanation
The ligands which induce pairing of the 3d electrons form inner orbital complexes exhibiting d2sp3 hybridisation. In the case of ligands which do not cause pairing of the 3d electrons, complexes are of the outer orbital type (sp3 d2 ), where the metal utilises 4d orbitals.
The magnetic moment of an ion in a complex is given by
⇒ \(\mu=\sqrt{n(n+2)}\) where n is the number of impaired electrons.
Example Find the expected magnetic moment for tetrahedral and square planar complexes of (2) and Co(2).
Solution
Ni(U) has d8 configuration and in tetrahedral complexes will have 2 unpaired electrons. The magnetic moment will be \(\mu=\sqrt{n(n+2)}=\sqrt{2} \times 4 \approx 2.8 \mathrm{BM}\)
Ni(2) in square planar complexes that have no unpaired electron and are diamagnetic.
Co(II) has d7 configuration and in tetrahedral complexes will have 3 impaired electrons, i.e., n = 3.
∴\(\mu=\sqrt{3(3+2)}=3.87 \mathrm{BM}\)
Co(2) in square-planar complexes will have one unpaired electron, i.e., n =1.
∴\(\mu=\sqrt{1(1+2)}=1.73 \mathrm{BM}\)
Example The spin-only magnetic moment of (./[NICIJ2 is 2.83 IIM. Predict the geometry of the complex
Solution
The coordination number of Ni(2) in the above complex is 4 and the geometry can be tetrahedral or square planar. the number of 3d orbitals of Ni(2) is 8. Square planar geometry involves dsp2 hybridisation in this case the d electron paired up and occupied will be diamagnetic.

If the geometry is tetrahedral, then the hybridisation is sp3 and there are two unpaired d electrons, making the complex paramagnetic.

In this case \(\mu=\sqrt{2(2+2)}=\sqrt{8}=2.83 \text { BM. }\)
Thus the complex is tetrahedral.
Colour In Coordination Compounds
Coordination compounds of transition metals display a range of colours. Absorption of light at a specific wavelength in the visible part of the electromagnetic spectrum causes the excitation of a d electron from a lower energy d orbital to a higher energy one.
The colour of the coordination entity observed is complementary to the wavelength absorbed. The relation between colours absorbed and light reflected is shown in.

Colour is associated with the electronic transition from a lower set of d orbitals to a higher set. The electron absorbs radiation in the visible range and undergoes this transition. When the electron returns to the original level the energy absorbed is emitted.
As the energy difference depends on the nature of the metal, the ligands and the oxidation number, it is obvious that the colour will be different in these cases.
For example, [Ni(H2O)6]2+ is green whereas [Ni(NH3)6]2+ is blue. The colour also varies with the oxidation number and coordination number of the metal. For example, [Cr(H2O)6]3+ and [Cr(H2O)6]2+ have different oxidation numbers.
The former is violet and the latter is blue. [Co(H2O)6]2+ and [CoCl4]2+ have different: coordination numbers. The former is pink and the latter is blue. The colour change is seen, since the value of •nergy difference depends on these factors.
Thus the crystal-field theory can successfully explain the colour of transition metal complexes.
The ion [Ti(H2O)6]3+ shows an absorption maximum at 498 runs and is violet in colour. This is an octahedral complex of Ti(m) which has a single d electron in a t2g level and the configuration may be represented as t21eg0. By absorbing energy corresponding to the blue-green region of the spectrum, the single d electron is promoted to the eg level and the configuration of the excited state is t2g0eg1.

This type of electronic transition is called a d-d transition and it arises due to the splitting of d orbitals. Crystal-field splitting will not occur in the absence of ligands and thus anhydrous CuSO4 is colourless.
The magnitude of crystal-field splitting depends on the ligand and thus the colour of complex changes with the ligand, for example [Cu(H2O)4]2+ is pale blue while [Cu(NH3)4]2+ is dark blue (almost purple).
When nickel(2) chloride is dissolved in water the complex [Ni(H2O)6]2+ is formed. Now, if an aqueous solution of a bidentate ligand, ethane-1, 2, -diamine (en), is added to the aqueous solution in the molar ratios1:1, 2: 1 and 3: 1 progressively, a colour change is observed at each step as follows.
⇒ \(\left[\mathrm{Ni}\left(\mathrm{H}_2 \mathrm{O}\right)_6\right]^{2+}+\mathrm{en}=\left[\mathrm{Ni}\left(\mathrm{H}_2 \mathrm{O}\right)_4(\mathrm{en})^{2+}+2 \mathrm{H}_2 \mathrm{O}\right.\)
pale blue
⇒ \(\left[\mathrm{Ni}\left(\mathrm{H}_2\mathrm{O}\right)_4(\mathrm{en})\right]^{2+}+\mathrm{en}=\underset{\text { purple }}{\left[\mathrm{Ni}\left(\mathrm{H}_2 \mathrm{O}\right)_2(\mathrm{en})_2\right]^{2+}}+\underset{2 \mathrm{H}_2 \mathrm{O}}{\mathrm{O}}\)
⇒ \(\left[\mathrm{Ni}\left(\mathrm{H}_2 \mathrm{O}\right)_2(\mathrm{en})_2\right]^{2+}+\text { en }=\underset{\text { violet }}{\left[\mathrm{Ni}(\mathrm{en})_3\right]^{2+}+2 \mathrm{H}_2 \mathrm{O}}\)
This shows how the colour of a complex changes under the influence of a ligand.
Limitations Of Crystal-Field Theory
We have seen that the crystal-field theory can successfully explain the formation of coordination compounds and their structures, colours and magnetic properties. However, it suffers from some inherent weaknesses. It cannot correlate the extent of crystal-field splitting with the charge on the ligand.
Anionic ligands having high charge density should cause greater splitting, but this is not generally so. Another defect is that it considers the metal-ligand interaction to be electrostatic and ignores the covalent character of the bond.
These drawbacks are taken care of in more advanced models like the molecular orbital theory and ligand-field theory, which are beyond the scope of this book.
Stability Of Coordination Compounds
A coordination compound generally does not dissociate appreciably in solution. The extent of dissociation depends upon the strength of the metal-ligand bond. The stability of a coordination compound is measured in terms of its stability constant.
A metal ion in an aqueous solution is hydrated. On adding a ligand to the solution, the water molecules are replaced by the ligand. This generally occurs in a step-wise manner as follows.
⇒ \(\begin{aligned}
& {\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_n\right]+\mathrm{L} \rightleftharpoons\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_{n-1} \mathrm{~L}\right]+\mathrm{H}_2 \mathrm{O}} \\
& {\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_{n-1} \mathrm{~L}\right]+\mathrm{L} \rightleftharpoons\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_{n-2} \mathrm{~L}_2\right]+\mathrm{H}_2 \mathrm{O}} \\
& {\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right) \mathrm{L}_{n-1}\right]+\mathrm{L} \rightleftharpoons\left[\mathrm{ML}_n\right]+\mathrm{H}_2 \mathrm{O}} \\
&\end{aligned}\)
The equilibrium constant for each step is referred to as the formation constant.
For example, the formation constant for the first step is
⇒ \(K_1=\frac{\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_{n-1} \mathrm{~L}\right]}{\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_n\right][\mathrm{L}]}\)
It may be noted that charges have been omitted and L is considered to be an unidentate ligand for the sake of simplicity.
The overall reaction may be written as
⇒ \(\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_n\right]+n \mathrm{~L} \rightleftharpoons\left[\mathrm{ML}_n\right]+n \mathrm{H}_2 \mathrm{O}\)
The stability constant or the equilibrium constant of the reaction is denoted by β.
⇒ \(\beta_n=\frac{\left[\mathrm{ML}_n\right]}{\left[\mathrm{M}\left(\mathrm{H}_2 \mathrm{O}\right)_n\right][\mathrm{L}]^n}\)
Note that the concentration of water is assumed to remain constant.
Tetrahedral crystal field splitting diagram and energy levels
It can be easily shown that βn = k1….k2……kn
In other words, the overall stability constant is the product of the stepwise stability constants.
The stepwise and overall stability constants are generally expressed as log K1, log K2, log βn etc. The stability of a coordination compound is directly proportional to its stability constant. Let us consider the stepwise formation of [Cd(CN)4]2-. The reactions are as follows.
⇒ \(\mathrm{Cd}^{2+}+\mathrm{CN}^{-} \rightleftharpoons \mathrm{Cd}(\mathrm{CN})^{+} ; K_1=\frac{\left[\mathrm{Cd}(\mathrm{CN})^{+}\right]}{\left[\mathrm{Cd}^{2+}\right]\left[\mathrm{CN}^{-}\right]}\)
⇒ \(\mathrm{Cd}(\mathrm{CN})^{+}+\mathrm{CN}^{-} \rightleftharpoons \mathrm{Cd}(\mathrm{CN})_2 ; K_2=\frac{\left[\mathrm{Cd}(\mathrm{CN})_2\right]}{\left[\mathrm{Cd}(\mathrm{CN})^{+}\right]\left[\mathrm{CN}^{-}\right]}\)
⇒ \(\mathrm{Cd}(\mathrm{CN})_2+\mathrm{CN}^{-} \rightleftharpoons \mathrm{Cd}(\mathrm{CN})_3^{-} ; K_3=\frac{\left[\mathrm{Cd}(\mathrm{CN})_3^{-}\right]}{\left[\mathrm{Cd}(\mathrm{CN})_2\right]\left[\mathrm{CN}^{-}\right]}\)
⇒ \(\mathrm{Cd}(\mathrm{CN})_3^{-}+\mathrm{CN}^{-} \rightleftharpoons \mathrm{Cd}(\mathrm{CN})_4^{2-} ; K_4=\frac{\left[\mathrm{Cd}(\mathrm{CN})_4^{2-}\right]}{\left[\mathrm{Cd}(\mathrm{CN})_3^{-}\right]\left[\mathrm{CN}^{-}\right]}\)
On adding Equations (i) to (iv), p4 =\(\beta_4=\frac{\left[\mathrm{Cd}(\mathrm{CN})_4^{2-}\right]}{\left[\mathrm{Cd}^{2+}\right]\left[\mathrm{CN}^{-}\right]^4}\)
The stability constant values are log K1=5.48, log K2 =5.12, log K3 = 4.63, log K4 = 3.65 and log β4 =18.8. There is a decrease in successive stability constants.
The reciprocal of the stability constant is referred to as the dissociation constant or instability constant and gives a measure of the extent of dissociation of the complex.
Example: The overall stability constant for the complex [Cu(NH3)4]2+ and the values of log β4, log K1 log K2 and log K3 respectively are as follows.
log β4=11-9, log K1 = 4.0, log K2 = 3.2 and log K3 = 2.7. Find the value of the stepwise stability constant, i.e., log K4.
Solution
Given
The overall stability constant for the complex [Cu(NH3)4]2+ and the values of log β4, log K1 log K2 and log K3 respectively are as follows.
Factors affecting crystal field splitting energ
log β4=11-9, log K1 = 4.0, log K2 = 3.2 and log K3 = 2.7.
We know that β4 = K1 x K2 x K3 x X4.
log β4 = log K1 + log K2 + log K3 + log K4
or log K4 = log β4 – (log K1 + log K2 + log K3 )
=11.9 -(4.0 + 3.2 + 2.7) = 2.0.
Example: The overall stability constant, pÿfor[Ni(NH3)6]2+ is 9.98 x107. Calculate the dissociation constant for the same.
Solution
Given
The overall stability constant, pÿfor[Ni(NH3)6]2+ is 9.98 x107.
The overall dissociation constant is the reciprocal of the overall stability constant, i.e. \(\frac{1}{\beta_6}\)
⇒ \(\frac{1}{\beta_6}=\frac{1}{9.98 \times 10^7}=1.002 \times 10^{-8} \text {. }\)