
Thermodynamics
This chapter deals with energy changes that take place during a chemical reaction. In any chemical reaction, the atoms of the reactants are rearranged to form the products. This involves the breaking and forming of bonds. You know that energy is required to break bonds and that it is released when bonds are formed. It should then be easy to understand that a chemical reaction involving the dissociation and formation of bonds must be accompanied by energy changes.
The energy change accompanying a reaction may appear in different forms. When fuels are burnt, for instance, energy appears as heat and light. The chemical reaction in a battery produces electrical energy. When a grenade explodes, the chemical reaction produces heat, light, sound and kinetic energy. Consider the following reactions.
| Class 11 Biology | Class 11 Chemistry |
| Class 11 Chemistry | Class 11 Physics |
| Class 11 Biology MCQs | Class 11 Physics MCQs |
| Class 11 Biology | Class 11 Physics Notes |
“Thermodynamics, definition, equations, and fundamental laws”
⇒ \(\mathrm{C}+\mathrm{O}_2\longrightarrow \mathrm{CO}_2 \text { + heat }\)
⇒ \(2 \mathrm{Mg}+\mathrm{O}_2\longrightarrow 2 \mathrm{MgO}+\text { heat }+ \text { light }\)
⇒ \(\mathrm{Zn}+\mathrm{CuSO}_4\longrightarrow \mathrm{ZnSO}_4+\mathrm{Cu}+\text { electrical energy }\)
In these reactions, there is a net release of energy. There are other reactions in which energy is absorbed. Take the case of electrolysis, for example. Here electrical energy is absorbed and the electrolyte splits into its components. Photosynthesis, on which almost all living organisms depend directly or indirectly, is another example of a reaction in which energy is absorbed.

Thermodynamics Some Definitions
Before we start a systematic study of the energy changes associated with chemical reactions, let us define a few basic terms used in any discussion of energetics or chemical thermodynamics.
System: A system is that part of the universe which we are interested in investigating. For example, if we are studying a particular reaction in a vessel, then the vessel, the reactants, and the products constitute the system.
Surroundings: Everything other than the system or the part of the universe other than the system is called the surroundings. In the example we have just considered, everything other than the vessel in which the reaction is taking place is called the surroundings. A system is separated from the surroundings by boundaries which may be real or imaginary. Further, boundaries may be considered part of either the system or the surroundings, depending upon convenience.
There are three types of systems: open system, closed system, and isolated system.
Open system A system which can exchange matter and energy with the surroundings is called an open system. For example, an open test tube in which a reaction is taking place is an open system. It can exchange heat with the surroundings and gaseous products can escape into the surroundings.
Closed system A dosed system can exchange energy with the surroundings but not mass. If a reaction occurs in a sealed bulb, which can exchange heat with the surroundings but not matter, the bulb with the reactants and products constitutes a closed system.
Isolated system An isolated system can neither exchange matter nor energy with the surroundings. A thermos flask filled with hot tea is an isolated system.
Intensive and extensive properties: The properties of any substance can be classified as intensive or extensive. Intensive properties are those independent of the size of the substance. For example, by doubling the size of the given sample of a substance, the temperature and pressure of the substance do not double or do not change. These are called Intensive properties, other examples being viscosity, density, and all other molar properties.
If the value of the property depends on the size of the substance, it is called an extensive property. On doubling the size, internal energy doubles, and hence it is an extensive property. Other examples include mass, volume, heat capacity, enthalpy, entropy, and free energy.
State functions: State functions are those parameters or measurable macroscopic properties of a system that describe its state. The state of a system is defined by specifying the values of certain number of macroscopic properties. The number depends on the nature of the system.
- The values of state functions or state variables depend only on the state of the system and not on how that state is readied. Take a gaseous system, for example. Its state is described by the state functions: temperature (T), pressure (p), and volume (IQ. To take a more specific case, consider 1 mol of CO at stp. Its volume will be 22.7 L irrespective of the method by which it is obtained.
- All state functions or state variables are not independent because equations of state exist between different state functions. For example, consider the equation of state for an ideal gas, pV-nRT. Here out of the four variables (p. V. n and T), only three can be independently varied.
- Thus once the values of the minimum number of state functions are fixed, others have definite values. The equilibrium state of a system is characterised by the definite values of state functions which do not change with time. Internal energy, enthalpy, and entropy are the state functions you will learn about in this chapter.
“Thermodynamics, first law, second law, third law, and equations“
The state of a system: As stated above, thermodynamic equilibrium exists in a system when the macroscopic properties or state functions do not change with time. If the equilibrium state is disturbed tire system again settles down to a new equilibrium state.
- If a particular state function does not have equal values in the system and its surroundings, exchange of matter or energy or both takes place between the thermodynamic system and its surroundings. After the interaction between system and its surroundings stops, the system attains a new equilibrium state with new values of state functions.
- The starting state of the system is referred to as the initial state and the state reached after interaction with the surroundings is the final state. When a thermodynamic system undergoes a change of state (from the initial to tire final), we say it has undergone a process. This implies that there will be a change in at least one of tire state functions of tire system during a process.
- A system can change from one state (initial state) to another (final state) through a number of paths. In other words, there can be a number of processes between an initial state and a final state. There are certain processes in which a particular state variable remains unchanged. Let us learn about these processes.
Isothermal If the temperature of the system remains constant during the change, the process is called isothermal.
Adiabatic If the system does not exchange heat with the surroundings, the process is called adiabatic.
Isobaric If the pressure of the system remains constant during the change, the process is called isobaric.
Isochoric If the volume of the system remains constant during the change, the process is called isochoric.
Reversible process In this process, the initial and the final states are connected through a succession of equilibrium states, i.e., at every state along the reversible path, there exists an equilibrium between the system and the surroundings. Such states are called quasi-equilibrium states.
Irreversible process The processes occurring in nature are generally irreversible. Their only difference from a reversible process is that equilibrium is not maintained during the transformation process.
Cyclic process The process that brings back a system to its original state after a series of changes is called a cyclic process.
Transference Of Energy
While describing open, closed, and isolated systems, we touched upon the idea of a system exchanging energy with the surroundings. There are two forms in which energy is exchanged between a system and the surroundings.
We often talk about heat flowing from a body at a higher temperature to one at a lower temperature. What we mean is that energy is exchanged between the two bodies because of a difference in temperature.
“Definition of thermodynamics, key equations, and basic laws
- So, one of the forms in which a system can exchange energy with the surroundings is heat. Energy is exchanged between a system and the surroundings in the form of heat when they are at different temperatures.
- The other mode of energy exchange between a system and the surroundings is energy. One example where energy is exchanged in this form is when the system and surroundings are at different pressures.
- Consider a gas enclosed in a cylinder that is fitted with a weightless, frictionless moving piston. If the gaseous system is at a higher pressure than the surroundings, the piston will move upwards (increasing the volume of the gas) until the pressure of the system and that of the surroundings become equal.
During the expansion of the gas, work is done by the system on the surroundings due to the pressure difference between the two. Here energy is transferred from the system to the surroundings as work.
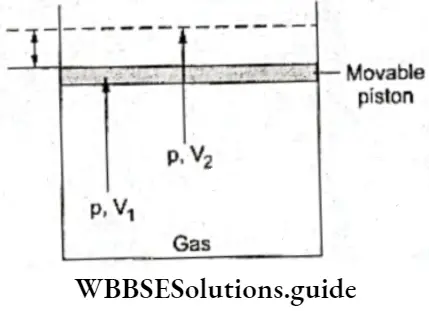
- If, on the other hand, the pressure of the system is lower than that of the surroundings, the piston is pushed down until the pressures become the same. In this case the volume of the gas decreases and work is done by the surroundings on the system.
- Once again there is figure, when the gas enclosed in a cylinder transference of energy in the form of work, but from the fitted with a piston is at a higher pressure than the surroundings to the system. By convention, work done on the surroundings, the piston moves until the pressures system is positive, while work done by the system is negative. equalize.
Remember that neither heat, nor work is a property of a system. They are path functions as they depend on how a change is brought about. A system does not possess a particular amount of work or heat. It does, however, have a particular amount of energy under a particular set of conditions. The SI unit of heat is the same as that of work, i.e., the joule. However, heat is sometimes expressed in calories or kilocalories.
1 calorie = 4.184 J.
1 J = 107 ergs.
Work
There are several kinds of work, such as mechanical work, electrical work, and chemical work. Work is said to be done if an object is moved by a force F through a distance d. The work done is given by
W = Fxd…….Equation 1
Some types of work encountered in chemistry are:
- Expansion work (which we shall study in detail) in the case of expansion or compression of a gas against a force surface expansion in case of liquids—these are examples of mechanical work
- When larger molecules are synthesized from smaller ones within living organisms—chemical work
- An ion moving in the presence of an electric field—electrical work.
The most common form of mechanical work encountered in chemistry is the expansion work associated with the expansion of a gas against pressure. It is also called p-V or pressure-volume work.
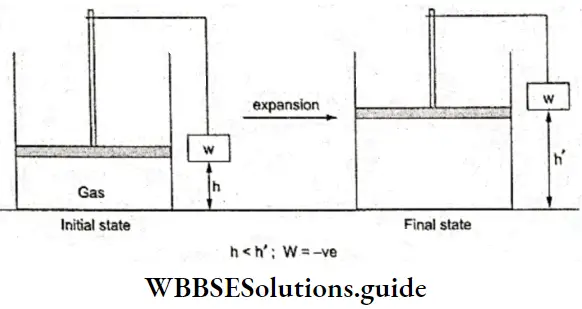
To understand the concept further, let us once again consider a Figure but with a weight attached to the piston. When the gas expands, the piston moves up and the weight is lifted up. We say that work is done by the system.
Let us now calculate the work done in terms of pressure and volume. If an external pressure pex is applied on the piston and the piston moves inwards by a distance dl, then the change in volume of the gas is given by
dV=A • dl …….(1)
where A is the area of the cross-section of the piston. The force on the piston is given as
F = pex • A …..(2)
Now, the work done in moving the piston by a distance dl against an opposing force of magnitude F is given by
dW = -Fdl …….(3)
(the negative sign is due to the opposing nature of the force).
Substituting (2) in (3)
dW = -pex • Adl
or dW = -pex dV
or simply W = -p Δ V …..Equation 2
- By the help of this equation, we can calculate the work done (or pressure/volume) during the expansion or compression of a gas. When expansion occurs ΔV in Equation 2 is positive, thereby resulting in a negative value for W.
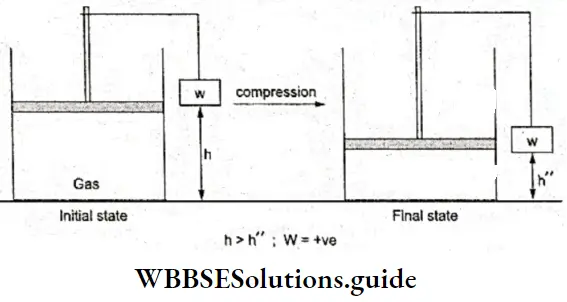
- Similarly, when compression occurs, weight is lowered in the surroundings and we say that work is done on the system. Since AV is negative now, W is positive.
- Thus we can say that work is done when there is a change in height of a weight in the surroundings.
If due to a change in state of a system, a weight is lifted in the surroundings, work is done by the system and if a weight is lowered in the surroundings, work is done on the system. Now W can also be given as W = mgh,
where m is the mass lifted, g is the acceleration due to gravity and h is the height through which the mass is lifted.
Thus, during a change in state of the system, we can learn about any work done by measuring the relevant quantities only in the surroundings.
Reversible expansion or compression of a gas: Suppose a gas confined in a cylinder with a movable piston is in equilibrium. This means that there is no change in state of the system. This is possible when the external pressure pex (pressure that is applied) is equal to the internal pressure, p, of the gas. A small change in pex can disturb the equilibrium and the gas may expand or compress, resulting in a change in the volume and hence the state of the system.
- When pex is increased infinitesimally (infinitesimal means negligibly small) then compression occurs and the volume of the gas decreases. Equilibrium with the surroundings is also established almost immediately. The system can also be restored to its original state by reducing the external pressure to the original value.
- By continuously increasing the external pressure, infinitesimally, the gas can be made to undergo a finite amount of compression. This is achieved by keeping different weights on the piston one after the other.
- In each step, however, the change is infinitesimal and can be reversed by an infinitesimal change in external pressure. Again, in each step equilibrium is immediately established.
- Thus we have a process which is carried out in several infinitesimally small steps and throughout the process the system is in equilibrium with its surroundings. This is a reversible process.
If the difference between the internal pressure and the external pressure is large then there is a considerable disturbance of equilibrium and an infinitesimal change in pex in the opposite direction will not bring back the system to its original state. Such a process is termed an irreversible process.
Calculation of work done in various cases
1. Work done in free expansion When the external pressure is zero (pex = 0), there is no opposing force on the piston and the gas can expand freely. This is called free expansion. This occurs when a gas expands in vacuum. Both the work done on the system and by the system are zero in this case.
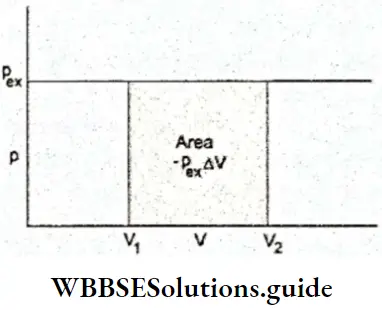
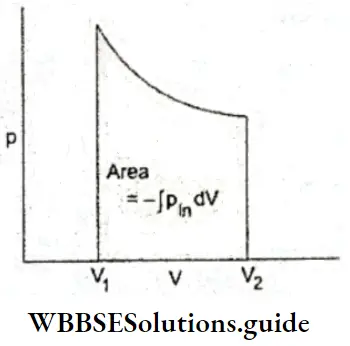
2. Work done in expansion against constant pressure If a constant external pressure is applied, the gas expands or compresses until the internal pressure becomes equal to the external pressure. The work done at every displacement dV is given by Equation 2. The total work done in the expansion from V1 to V2 is the sum of all such contributions. It is obtained by finding the area under the p-V plot, i.e., by integrating Equation.
∴ \(W=-\int_{V_1}^{V_2} p_{e x} d V\) ….. (1)
= \(-p_{e x} \int_{V_1}^{V_2} d V=-p_{e x}\left(V_2-V_1\right)\)
∴ \(W=-p_{e x} \Delta V\)
In case of expansion, ΔV is positive and W turns out to be negative. Therefore, work is done by the system.
If ΔV is negative as in case of compression, W is positive, i.e., work is done on the system.
3. Work done in reversible expansion In reversible expansion, the difference between external and internal pressure is infinitesimally small. With very small changes in pressure, the process of expansion and compression may be reversed. If the expansion occurs in several stages, at each stage, it is ensured that the external pressure is only infinitesimally different (less in case of expansion and more in case of compression) from the internal pressure of the gas.
Since \(p_{e x}=p_{\text {in }}\) we may replace pex in Equation 2 by pin
dW = -pin dV.
This is the work done at every stage of the expansion. The total work is obtained by summing up all such infinitesimal contributions (again) given by the area under the p-V plot.
⇒ \(\int d W=-\int p_{\mathrm{ma}} d V\)
∴ \(W_{\mathrm{rev}}=-\int p_{\mathrm{in}} d V\)
The subscript ‘rev’ indicates the reversible nature of the process.
If it is assumed that the gas behaves ideally then pin be replaced by nRT/v using the ideal gas equation.
Therefore \(W_{\mathrm{rev}}=-\int_{V_1}^{V_2} \frac{n R T}{V} d V\)
If the process is carried out at a constant temperature (isothermally), T may be taken out of the integral sign.
“Thermodynamics, important formulas, equations, and principles”
⇒ \(W_{r e v}=-n R T \int_{V_1}^{V_1} \frac{d V}{V}\)
= \(-n R T|\ln V|_{V_1}^{V_2}\)
or, \(W_{n e v}=-n R T \ln \left(\frac{V_2}{V_1}\right)=-2303 n R T \log \frac{V_2}{V_1}\),
where V1 and V2 are the initial and final volumes respectively.
Internal Energy
The energy stored within a system (say chemical system) is its internal energy or intrinsic energy. Under a particular set of conditions, a thermodynamic system has a definite amount of internal energy.
- In other words, the internal energy of a system, generally represented as U, depends on the nature, amount, temperature, and pressure of the system. This energy possessed by a system is due to the different types of energy that its atoms or molecules have.
- It is, in fact, the sum of the energies of the elementary particles of the system, i.e., the sum of their potential and kinetic energies and the bond energy between constituting atoms.
- Instead of saying that a particular system under a particular set of conditions possesses a definite amount of internal energy, we could have said that internal energy is a state function, which means that it depends only on the initial and final state of the system and is independent of the way (path) the change takes place.
However, unlike other state functions, like pressure and temperature, it is not possible to determine the value of internal energy. This is because it is not possible to determine with exactness the values of the components of internal energy. Note that this matters little, because in the study of chemical processes, what we are really interested in knowing is the clumge in internal energy, ΔU (delta U).
ΔU = U2-U1
where U2 is the internal energy of the system at the end of the process being studied and is the internal energy of the system at the start of the process. For a chemical reaction
ΔU = Up-Ur,
where U2 is the internal energy of the products and Ur is that of the reactants. Obviously, All is a state function since Up and Ur are state functions.
So, how do we measure ΔU or the change in internal energy associated with a chemical process? Suppose we carry out a chemical reaction in such a way that the system remains at the same temperature, there is no work done on the system and it does no work on the surroundings.
- Whatever the change in the internal energy of the system in the course of the reaction must be equal to the energy exchanged by the system with the surroundings in the form of heat.
- Why? This in fact follows from the law of conservation of energy, which we will discuss subsequently, for the moment, imagine that the change in internal energy of the system is negative, i.e., Up < Ur.
- What happens to this energy which the system has apparently lost? It cannot just vanish because that is against the law of conservation of energy. We have assumed that the system does not work and nor is any work done on it (i.e., volume remains constant), so the energy cannot be exchanged with the surroundings in the form of work.
- Since the temperature is constant, the energy must be exchanged with the surroundings in the form of heat.
- The change in the internal energy associated with a chemical reaction is, thus, determined by letting the reaction occur at constant temperature and constant volume and measuring the heat exchanged with the surroundings.
Law Of Conservation Of Energy
This is one of those common-sense laws which seem very obvious but have far-reaching implications. It states that energy can neither be created nor destroyed, though it may change from one form to another.
In the context of the system and the surroundings that we have been discussing, this means that the total energy of the system and the surroundings (i.e., the universe) remains constant.
- More specifically, it means that during a chemical reaction, energy may be absorbed or released, but the total energy of the reaction system and the surroundings remains constant.
- The law of conservation of energy is also known as the first law of thermodynamics. It is of relevance not only to chemists but also to physicists, engineers, and others who deal with the conversion of energy from one form into another.
The first law of thermodynamics being identical to the law of conservation of energy can be stated as:
- Energy can neither be created nor destroyed although it can be changed from one form to another.
- The energy of an isolated system is constant.
Consider the universe, which is an isolated system. Tire energy of the universe is conserved. Inside the universe, energy can be transferred from one part to another or it can be converted from one form to another, but it can neither be created nor destroyed.
Let us see if we can express this law mathematically. If the internal energy of a particular system is U1 and it absorbs a certain amount of heat q, then its internal energy will become U1 + q. Now suppose W amount of work is done on the system, then U2, the final energy of the system is
U2 =U1 +q + W
or ΔU = U2 – U1 = q + W….. Equation 3
(If W is the work done on the system, -W is the work done by the system.)
- This means the change in the internal energy of the system is equal to the sum of the heat absorbed by the system and the work done on it.
- One can generalise this to say the change in the internal energy of the system is the sum of the heat exchanged by the system (with the surroundings) and the work done on or by the system. Equation 3 is the mathematical statement of the first law of thermodynamics.
If we now substitute for W in Equation 3, the expression for change in internal energy (when only pressure-volume type of work is done) becomes
ΔU = q – pΔV …. Equation 4
Now, if there is no change in the volume of the system then W = 0, i.e., W = -∫pdV = 0.
We can easily see that if there is no change in volume during a reaction, Equation 4 becomes ΔU = qv,
where qv is the heat exchanged (evolved or absorbed) by the reaction system at constant volume. When heat is absorbed by the system, q is positive and when heat is evolved by the system, q is negative. This is exactly what we said in the previous section.
The change in the internal energy of a system during a chemical reaction is equal to the heat exchanged with the surroundings, provided the volume and temperature of the system remain constant.
For any isothermal expansion of an ideal gas the total energy remains the same, and q = -W. In other words, the internal energy remains constant (ΔU = 0) when an ideal gas expands isothermally.
“Thermodynamics, laws of energy, entropy, and enthalpy equations”
Thus for an isothermal irreversible change, q = -W = pex ΔV
and for an isothermal reversible change, q = \(-W=n R T \ln \frac{V_2}{V_1}\)
= \(-2.303 n R T \log \frac{V_2}{V_1}\),
where V1 and V2 are the initial and final volumes respectively.
For an adiabatic change, q = 0,
∴ ΔU = Wadiabstic (from Equation 3).
Enthalpy
So far we have considered reactions taking place at constant volume and temperature, and the energy changes associated with such reactions. But the reactions we carry out in the laboratory normally occur in open vessels (beakers or test tubes, say).
Obviously, the volume of such a reaction system does not remain constant. However, the pressure does, since such a system, open to the atmosphere, is at atmospheric pressure and this pressure is more or less constant.
- The heat exchanged with the surroundings by a reaction system in the course of a reaction is equal to the change in the internal energy of the system if the temperature and volume are constant.
- What about the heat exchanged with the surroundings by a reaction system at constant pressure and not maintained at constant volume? It should be different, but why and how?
- Let us consider a reaction in which the volume of the system increases. An increase in volume can occur when the system does some work against the atmospheric pressure and energy is required for this work.
- Thus, in this case, the heat exchanged with the surroundings would be lower than that for a system at constant volume and temperature.
- On the other hand, if the reaction involves a decrease in the volume of the system, work would have to be done on the system by tire surroundings. The heat exchanged would then be greater than that exchanged by a system at constant volume.
- Thus, when a reaction proceeds at constant pressure and temperature, the heat exchanged by the system with tire surroundings is not equal to the change in the internal energy of the system.
In other words, the change in the internal energy of the system is not the only contributing factor to the total energy change associated with the reaction. Tire total energy change includes change in energy due to the pressure-volume type of work done by or on tire system.
Enthalpy or heat content is a property or state function introduced to take care of the energy changes associated with tire kind of system we have just been discussing. It is denoted by H and expressed mathematically as follows.
H = U + pV, …… Equation 5
where U is the internal energy, p the pressure, and V the volume of the system. The enthalpy of a system (substance) can be defined as the total energy associated with it, i.e., its internal energy and the energy due to factors such as pressure-volume conditions. The enthalpy of a substance depends on its state (temperature, pressure, etc.).
- Enthalpy changes are normally expressed with the substances in their standard states. The standard state of a pure substance is the pure form at a pressure of 1 bar and a specified temperature, the conventional temperature being 298.15 K.
- Enthalpy change in the standard state is called the standard enthalpy change. It is denoted by \(\Delta H^ominus\), where the superscript \(\ominus\) denotes standard conditions.
The molar enthalpy of a substance Hm = H/n (where n is the number of moles) is an intensive property and Hm = Um + pVm. When enthalpy changes have to be compared, the states of the systems must be identical.
Enthalpy change: As in the case of internal energy, the absolute value of the enthalpy of a system cannot be determined. What we are interested in knowing is the enthalpy change associated with a process. The total change in the energy of a system during a reaction at constant pressure is the enthalpy change, denoted by ΔH.
∴ \(\Delta H=H_{\text {products }}-H_{\text {reactants }}\)
= \(\left(U_p+p V_p\right)-\left(U_r+p V_r\right)\)
= \(\Delta U+\Delta p V\),
where Up = internal energy of products,
Vp =volume of products,
Ur = internal energy of reactants, and Vt = volume of reactants.
Since p is constant, ΔH = ΔU + pΔV…….. Equation 6
The enthalpy change during a process is the sum of the change in internal energy and the pressure-volume work done.
Suppose the heat exchanged by a system with the surroundings during a reaction at constant pressure and temperature is qp. Suppose also that the change in internal energy of the system is ΔU and the work done on the system is W. Then
qp = ΔU-W = ΔU – (-pΔV)
= ΔU + pΔV
= ΔH. (ΔU + pΔV = ΔH)
- The heat exchanged by a system with the surroundings during a reaction at constant pressure and temperature is the enthalpy change associated with the reaction. In practice, the enthalpy change associated with a reaction is determined by insulating the system and allowing the heat of the reaction to alter its temperature.
- The amount of heat required to be supplied to or taken away from the system to let its temperature go back to the original value is then calculated to obtain the enthalpy change.
Significance of ΔU and ΔH: It is understood that the amount of heat exchanged with the surroundings for a reaction at a constant pressure (ΔH) is different from that exchanged at constant volume and temperature (ΔU).
The difference is not significant for solid or liquid systems but it does matter when gases are involved. Let us consider a gaseous reaction, where Vr is the total volume of the gaseous reactants Vp is the total volume of gaseous products, nr, is the number of moles of reactants and nr is the number of moles of products, all at constant pressure and temperature.
By the ideal gas law,
∴ \(\quad p V_t=n_r R T\)
and \(p V_p=n_p R T\)
Thus, \(p V_p-p V_r=\left(n_p-n_r\right) R T\)
or \(\quad p\left(V_p-V_r\right)=\left(n_p-n_r\right) R T\)
or, \(\quad p \Delta V=\Delta n_g R T\).
Here, Δng is the difference between the number of moles of the gaseous products and that of the gaseous reactants.
From the definition of enthalpy, it follows that ΔH = ΔU + pΔV.
Therefore, ΔH = ΔU + ΔngRT.
The above equation is useful in situations where one of the two quantities ΔH and ΔU is known and the other has to be calculated. ΔH and ΔU differ significantly for processes involving gases. For solids and liquids, pVp is only slightly different from pVx.
Source of enthalpy change: You may be wondering what exactly leads to enthalpy change during a reaction. Any reaction involves the breaking and forming of bonds.
- Energy is released when bonds are broken and required for the formation of bonds. Simply put, the net change in energy due to the breaking and forming of bonds is the enthalpy change of the reaction.
- Of course, for the net change in energy to be termed enthalpy change, the reaction must take place at constant pressure and temperature. The simplest case is that of a gaseous system in which gas A reacts with gas B to form gas C.
If the reaction involves solutions, the interactions (hence energy changes) between the solvents and the reactants and products have to be considered. For liquid or solid reactants, we have to also consider the interactions between the molecules.
Enthalpy change(for a gaseous system at constant pressure) = (energy required to break bonds) – (energy released in formation of bonds).
Let us consider the reaction between hydrogen gas and chlorine gas to form HCl gas. Energy is required to break tire H—H and Cl—Cl bonds and released when H—Cl bonds are formed.
∴ \(\mathrm{H}_2(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{HCl}(\mathrm{g})\)
The energy required to break H—H bonds = 436 kJ mol-1.
Energy required to break Cl—Cl bonds = 242 kJ mol-1.
Energy released in the formation of two H—Cl bonds = 2 x 431 kj mol-1.
Enthalpy change (ΔH) = 436 + 242 – 2 x 431 = -184 kJ.
The negative value of ΔH indicates a decrease in the enthalpy of the system.
The heat evolved in this reaction is 184 kJ mol-1.
Exothermic And Endothermic Reactions
In our discussion of the energy changes associated with chemical reactions so far, we have only mentioned that energy is released in some reactions and absorbed in others. Reactions in which energy is released are called exothermic while those in which energy is absorbed are called endothermic.
Exothermic reactions: While writing an exothermic reaction, the heat evolved is indicated on the right side, after the products.
∴ \(\mathrm{N}_2+3 \mathrm{H}_2 \longrightarrow 2 \mathrm{NH}_3+93.7 \mathrm{~kJ}\)
- If an exothermic reaction is carried out at constant volume and temperature, the heat evolved (qν) is (numerically) equal to the change in internal energy (ΔU). In such a reaction the internal energy of the products (Up) is less than the internal energy of the reactants (Ur) and ΔU is negative.
- If an exothermic reaction occurs at constant pressure and temperature, the heat evolved is (numerically) equal to the change in enthalpy (ΔH). The enthalpy of the products is less than the enthalpy of the reactants, i.e., ΔH is negative.
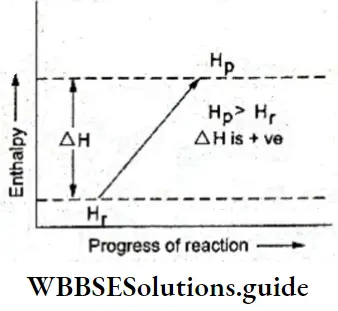

Endothermic reactions: In the case of an endothermic reaction, the heat absorbed can be indicated along with reactants or with the products. Obviously, if it is shown on the left side of the equation, it will have a positive sign, and if it is shown on the right side, it will carry a negative sign.
∴ \(\mathrm{N}_2+\mathrm{O}_2+180.5 \mathrm{~kJ} \longrightarrow 2 \mathrm{NO}\)
or, \(\mathrm{N}_2+\mathrm{O}_2 \longrightarrow 2 \mathrm{NO}-180.5 \mathrm{~kJ}\)
When an endothermic reaction occurs at constant temperature and constant volume, the heat absorbed is (numerically) equal to the change in the internal energy of the system. The internal energy of the products (Up) is greater than the internal energy of the reactants (Ur) and ΔU is positive.
When an endothermic reaction proceeds at constant pressure and temperature, the heat absorbed is (numerically) equal to the change in enthalpy of the system. The enthalpy of the products (Hp) is greater than the enthalpy of the reactants (Hr) and ΔH is positive.
Thermochemical Equations
You already know how to write a chemical equation. When a chemical equation not only indicates the quantities and physical states of the reactants and products involved, but also the change in enthalpy during a reaction, it is called a thermochemical equation. Fractional coefficients may also be used in such an equation.
⇒ \(2 \mathrm{H}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{H}_2 \mathrm{O}(\mathrm{l}) ; \quad \Delta H=572 \mathrm{~kJ}\)
Certain conventions must be followed while writing a thermochemical equation. These conventions are listed below.
1. The heat evolved or absorbed is indicated as in the equation above, in terms of ΔH. Remember that ΔH is negative for exothermic reactions and positive for endothermic reactions.
2. The numerical value of ΔH corresponds to the reaction as written. In the absence of any information, it is assumed that a certain value of ΔH is due to the number of moles of reactants that combine as indicated by the chemical equation. Thus ΔH is expressed in kJ mol-1.
3. The value of standard enthalpy (\(\Delta H^{\ominus}\)) in a thermochemical equation corresponds to the standard state of the substances involved in the reaction. The term ‘standard state of a substance’ refers to the pure substance at exactly 1 bar pressure.
4. The coefficients of the various substances represent the number of moles of the substances involved in the reaction and the value of ΔH corresponds to these coefficients. It stands to reason, therefore, that if the coefficients are multiplied or divided by some factor, so must ΔH be. Consider the following example.
⇒ \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \quad \Delta H=-242 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
“Thermodynamics, Zeroth law, first law, second law, and applications”
If the coefficients are multiplied by 2, ΔH must also be multiplied by 2.
⇒ \(2 \mathrm{H}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \quad \Delta \mathrm{H}=-(242 \times 2) \mathrm{kJ} \mathrm{mol}^{-1}\)
When the reaction is reversed, the sign of AH is reversed but its magnitude remains the same.
\(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{HI}(\mathrm{g})\Delta H=+53.9 \mathrm{~kJ} \mathrm{~mol}^{-1}\)⇒ \(2 \mathrm{HI}(\mathrm{g}) \longrightarrow \mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \Delta H=-53.9 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
5. The physical states of the reactants and products have to be indicated because ΔH changes with the physical state. The following example will make this clear.
⇒ \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g})\Delta H=-242 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
⇒ \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l})\Delta H=-286 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Enthalpy Of Reactions
The enthalpy change accompanying a reaction is called reaction enthalpy and is denoted by ΔrH. The standard enthalpy of reaction (ΔrHΘ) is the change in enthalpy per unit amount of the reaction when the reactants in their standard states change to products in their standard states.
By standard states we mean reference state, i.e., the most stable state of aggregation. For example the reference state of H2 is pure gas at 1 bar and that of CaCO3 is pure solid at 1 bar. By convention the standard states are reported at 298 K. There are several factors that determine the value of enthalpy of a reaction.
Quantities of reactants The amount of heat absorbed or released as also the enthalpy of a reaction depends on the quantities of reactants involved. This should be pretty easy to understand. If you bum 1 kg of coal it will produce less heat than if you bum 10 kg of coal.
The standard enthalpy change for the reaction \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \quad \Delta_{\mathrm{r}} \mathrm{H}^{\ominus}=-393.5 \mathrm{~kJ} \mathrm{~mol}^{-1}\) has the units of kJ mol-1. Per mole refers to the reaction as written above.
Let us take another example.
⇒ \(2 \mathrm{CH}_3 \mathrm{OH}(\mathrm{l})+3 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}_2(\mathrm{~g})+4 \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
∴ \(\Delta_{\mathrm{r}} H^{\ominus}\) for this reaction is -1276.9 kj mol-1.
However, if the reaction is written with different stoichiometric coefficients, i.e., \(\mathrm{CH}_3 \mathrm{OH}(\mathrm{l})+\frac{3}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g})+2 \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
∴ \(\Delta_{\mathrm{r}} H^{\ominus}\) is -638.4 kJ mol-1, ‘per mole’ referring to the reaction as written here.
Physical State of Reactants and products The change of state of a substance involves heat changes, so the enthalpy of a reaction depends on the physical states of the reactants and products. We have already discussed the case of the formation of liquid water and gaseous water vapour from gaseous hydrogen and oxygen.
Allotropic forms The enthalpy of a reaction also depends on the allotropic forms of the substances involved in the reaction.
⇒ \(\mathrm{C} \text { (diamond) }+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \Delta H^{\ominus}=-395.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
⇒ \(\mathrm{C} \text { (graphite) }+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \Delta H^{\ominus}=-393.5 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Temperature The enthalpy of a reaction depends on the temperature at which the reaction proceeds. The value of enthalpy indicated in a thermochemical equation is generally the value at 298 K.
Constant pressure or volume As we have discussed already, the enthalpy of a reaction at a particular temperature depends on whether the reaction occurs at constant pressure or constant volume. From our discussion so far
ΔH = ΔU + pΔV.
But ΔH = qp and ΔU = qv.
∴ qp = qv + pΔV
Hess’s Law
The enthalpy of a substance is a stale function, independent of the method by which the substance is made. The enthalpy change in a reaction is the difference between the enthalpies of the products and the reactants.
- Both of these are consequences of the law of conservation of energy. Another consequence of the law is what is known as
Hess’s law of constant heat summation, which states that the enthalpy change in a chemical or physical process is independent of the path taken or the manner in which the change is brought about. - In the context of a chemical reaction, the law can be stated as follows. “The amount of heat absorbed or released during a reaction is the same whether the reaction proceeds in a single step or through several steps.”
Though G H Hess (a Russian chemist) came up with this law as a result of experimental observations, if you think a little you will realise that it follows from, or is a corollary of, the law of conservation of energy. How? Suppose a reactant A is converted into a product D and the heat evolved in the process is q.
- Now suppose the same reactant A changes first to B, then B is converted to C and finally, C changes to D and the heat evolved in the three steps is q1, q2, and q3. Let q1 + q2 + q3 = q′. According to Hess’s law q = q’. If Hess’s law were not true, either q> q’ or q < q’. Let us consider the possibility that q >q’.
- Then the heat evolved in converting A to D in a single step would be greater than the heat absorbed when D is converted to A in three steps. This would lead to a situation in which (q – q‘) of heat would be ‘created’ after the completion of the cyclic process and the law of conservation of energy would be violated.
Hess’s law can be stated as follows: The enthalpy change for a reaction that is the sum of two or more other reactions is equal to the sum of the enthalpy changes of the constituent reactions.
Let us consider two ways in which carbon dioxide may be produced from carbon and oxygen. Either carbon dioxide can be produced directly by the combustion of carbon, or carbon can first be converted to carbon monoxide, which can then be converted to carbon dioxide.
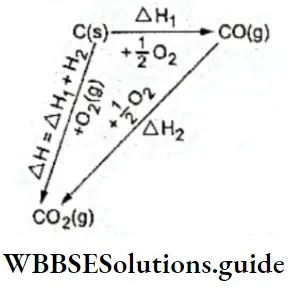
⇒ \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \Delta_{\mathrm{r}} H^{\ominus}=-394 \mathrm{~kJ}\) …. (1)
⇒ \(\mathrm{C}(\mathrm{s})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}(\mathrm{g}) \Delta_{\mathrm{r}} H_1^{\ominus}=-110.5 \mathrm{~kJ}\) …. (2)
⇒ \(\mathrm{CO}(\mathrm{g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \Delta_{\mathrm{r}} H_2^{\ominus}=-283.5 \mathrm{~kJ}\) …..(3)
Adding the enthalpy changes in (2) and (3) you will get -394 kJ mol-1, which is the same as the enthalpy change in (1). Do you see now why the law is called Hess’s law of constant heat summation?
Application of Hess’s law: Hess’s law is very useful in determining enthalpy changes in reactions for which it is not possible to experimentally determine enthalpy changes. It follows from Hess’s law that thermochemical equations can be added, subtracted, multiplied, or divided like algebraic equations.
Let us again consider the reaction between carbon and oxygen. The principal product of this reaction is CO2 but CO may also be produced due to insufficient oxygen and this may then form CO2. It is difficult to measure directly the enthalpy of combustion of carbon to carbon monoxide, CO. Let us assume it is unknown and determine its values by applying Hess’s law.
Reaction for which enthalpy is to be found: \(2 \mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}(\mathrm{g})\) …(1)
Reactions for which data is available: \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g})\) \(\Delta_{\mathrm{r}} H^\ominus=-393.5 \mathrm{~kJ}\) …. (2)
⇒ \(2 \mathrm{CO}(\mathrm{g})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}_2(\mathrm{~g})\) \(\Delta_{\mathrm{r}} H^{\ominus}=-566.0 \mathrm{~kJ}\) ….. (3)
How do we use these equations (with data) to get equation (1)? We need C and O2, on the LHS and CO on the RHS. In Equation (2) C and O2 are already on the LHS but the coefficient of C is 1 while we require 2 (from Equation (1)).
Hence, by multiplying Equation (2) by 2, we get
⇒ \(2 \mathrm{C}(\mathrm{s})+2 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}_2(\mathrm{~g}) \quad \Delta_{\mathrm{r}} H^{\ominus}=2 \times(-393.5) \mathrm{kJ}\) ….(4)
[If the equation is multiplied by 2, \(\Delta_r H^{\ominus}\)must also be multiplied by 2.]
Next CO must appear on the RHS. Reversing Equation (3) to get CO on RHS, we get
⇒ \(2 \mathrm{CO}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}(\mathrm{g})+\mathrm{O}_2(\mathrm{~g}) \quad \Delta_{\mathrm{r}} H^{\ominus}=-(-566)=+566 \mathrm{~kJ}\) …. (5)
[If the reaction is reversed, the sign of ΔH must also be reversed.]
Now adding reactions (4) and (5)
∴ \(2 \mathrm{C}(\mathrm{s})+2 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}_2(\mathrm{~g}) \quad \Delta_{\mathrm{r}} H^{\ominus}=-787.0 \mathrm{~kJ} \\
\begin{array}{lll}
2 \mathrm{CO}_2(\mathrm{~g}) & \longrightarrow 2 \mathrm{CO}(\mathrm{g})+\mathrm{O}_2(\mathrm{~g}) \Delta_{\mathrm{r}} H^{\ominus}=566 \mathrm{~kJ} \\
\hline 2 \mathrm{C}+\mathrm{O}_2 \longrightarrow 2 \mathrm{CO} & \Delta H^{\ominus}=-787+566=-221 \mathrm{~kJ}
\end{array}\)

Enthalpy Of Formation
The enthalpy or heat of the formation of a substance is the heat change accompanying the formation of 1 mol of the substance from its constituent elements and is usually written as ΔtH. If all the substances involved in the reaction are in the standard state, the heat change is referred to as the standard heat of formation and is denoted by \(\quad \Delta_{\mathrm{f}} H^{\ominus}\) Consider the following equations.
∴ \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \quad \Delta_{\mathrm{f}} H^{\ominus}=-393.5 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
This means the heat liberated in the formation of 1 mol of gaseous carbon dioxide from its constituent elements is 393.5 kJ or that the enthalpy of formation of carbon dioxide is 393.5 kJ mol-1.
∴ \(6 \mathrm{C}(\mathrm{s})+6 \mathrm{H}_2(\mathrm{~g})+3 \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{C}_6 \mathrm{H}_{12} \mathrm{O}_6(\mathrm{~s}) \quad \Delta_{\mathrm{f}} H^{\ominus}=-1169 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
When 1 mol of glucose is formed from its constituent elements in their standard states, the heat liberated is 1169 kJ, or the standard molar enthalpy of formation of glucose is 1169 kJ mol-1 (‘molar’ referring to the formation of one mole of the substance).
By convention, the standard enthalpy of formation of elements is zero at all temperatures. However, when an element changes its state, the standard enthalpy of formation is not zero.
Usefulness of \(\Delta_{\mathrm{f}} H^{\ominus}\).
If you know the standard enthalpies of formation of the reactants and products of a chemical reaction, you can easily calculate the standard enthalpy change for that reaction. The difference between the sum of the enthalpies of formation of the products and the sum of the enthalpies of formation of the reactants is the enthalpy change associated with the reaction.
Standard enthalpy = \(\left(\begin{array}{c}
\text { sum of standard heats of } \\
\text { formation of products }
\end{array}\right)-\left(\begin{array}{c}
\text { sum of standard heats of } \\
\text { formation of reactants }
\end{array}\right)\)
∴ \(\Delta_r H^{\ominus}=\Sigma \Delta_f H^{\ominus} \text { (products) }-\Sigma \Delta_f H^{\ominus} \text { (reactants) }\)
Example 1. Calculate the enthalpy change for the following reaction. \(\mathrm{CO}_2(\mathrm{~g})+\mathrm{H}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}(\mathrm{g})+\mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
Given that \(\Delta_t H^{\ominus}\) for CO2(g), CO(g) and H2O(g) are -393.5 kJ mol-1, -111.3 kJ mol-1, and -241.8 kJ mol-1 respectively.
Solution:
Given that \(\Delta_t H^{\ominus}\) for CO2(g), CO(g) and H2O(g) are -393.5 kJ mol-1, -111.3 kJ mol-1, and -241.8 kJ mol-1 respectively.
⇒ \(\Delta_{\mathrm{r}} H^{\ominus}=\Sigma \Delta_{\mathrm{f}} H^{\ominus} \text { (products) }-\Delta_{\mathrm{f}} H^{\ominus} \text { (reactants) }\)
= \(\left[\Delta_f H^\ominus(\mathrm{CO})+\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{H}_2 \mathrm{O}\right)\right]-\left[\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{CO}_2\right)+\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{H}_2\right)\right]\)
=[(-1113) + (-2418)]-[(-393.5) + (0)] =- 353.1 + 393.5 = 40.4 kJ mol-1.
Example 2. Calculate the enthalpy change for the following reaction.
\(\mathrm{CCl}_4(\mathrm{~g})+2 \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g})+4 \mathrm{HCl}(\mathrm{g})_{;} \Delta_{\mathrm{r}} H^{\ominus}=-41.4 \mathrm{kcal}\)
Given that,
⇒ \(\Delta_{\mathrm{f}} \mathrm{H}^{\ominus} \text { for } \mathrm{CCl}_4(\mathrm{~g}), \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \text { and } \mathrm{CO}_2(\mathrm{~g}) \text { are }\) -25.5 k cal mol-1,-57.8 k cal mol-1 and -94.1k cal mol-1 respectively.
Solution:
Given that,
⇒ \(\Delta_{\mathrm{f}} \mathrm{H}^{\ominus} \text { for } \mathrm{CCl}_4(\mathrm{~g}), \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \text { and } \mathrm{CO}_2(\mathrm{~g}) \text { are }\) -25.5 k cal mol-1,-57.8 k cal mol-1 and -94.1k cal mol-1 respectively.
⇒ \(\Delta_{\mathrm{r}} H^{\ominus} =\left[\Sigma \Delta_{\mathrm{f}} H^{\ominus}(\text { products })\right]-\left[\Sigma \Delta_{\mathrm{f}} H^{\ominus}(\text { reactants })\right]\)
= \(\left[\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{CO}_2\right)+4 \times \Delta_{\mathrm{f}} H^{\ominus}(\mathrm{HCl})\right]-\left[\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{CCl}_4\right)+2 \times \Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{H}_2 \mathrm{O}\right)\right]\)
-41.4 = \(\left[(-94.1)+4 \Delta_{\mathrm{f}} H^{\ominus}(\mathrm{HCl})\right]-[(-25.5)+(2 \times-57.8)]\)
or -41.4 = \(-94.1+4 \Delta_{\mathrm{f}} H^{\ominus}(\mathrm{HCl})+141.1\)
∴ \(4 \times \Delta_{\mathrm{f}} H^{\ominus}(\mathrm{HCl})=-41.4-141.1+94.1=-88.4\).
∴ \(\Delta_f H^{\ominus}(\mathrm{HCl})=\frac{-88.4}{4}=-22.1 \mathrm{kcal} \mathrm{mol}^{-1}\).
In our discussion so far, we have not really distinguished between various types of reactions or processes such as combustion, neutralization, and change of state. When we have used the term enthalpy of a reaction, we have used it to refer to any chemical process or reaction. The enthalpies associated with different types of reactions or processes actually go by different names.
Enthalpy of combustion: The enthalpy or heat of combustion of a substance is the heat change accompanying the complete combustion of 1 mol of the substance in (excess) oxygen or air. The heat of combustion of carbon, for instance, is -393.5 kJ mol-1.
⇒ \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) ; \quad \Delta_{\mathrm{c}} H^{\ominus}=-393.5 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Standard enthalpy of combustion is defined as the enthalpy per mol of a substance, when all the reactants and products are in their standard states at the specified temperature.
Remember that the heat of combustion of a substance is the heat evolved when 1 mol of the substance is completely burnt or oxidised. Carbon is converted to carbon monoxide according to the following reaction.
⇒ \(\mathrm{C}(\mathrm{s})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}(\mathrm{g}) ; \quad \Delta_c H^{\ominus}=-110.5 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
But -110.5 kJ mol-1 is not the heat of combustion of carbon.
Remember also that combustion reactions are always accompanied by the evolution of heat, so the heat of combustion is always negative.
Calorific value Combustion reactions are very important for us. We bum fuels to meet our energy requirements. We use the energy released by the oxidation of food to survive. The calorific value of a fuel or food is the amount of heat (in calories) released when 1 mol of the fuel or food is burnt or oxidised completely.
Example 1. A cylinder of cooking gas contains 11 kg of butane. The thermochemical equation for the combustion of butane is \(\mathrm{C}_4 \mathrm{H}_{10}(\mathrm{~g})+\frac{13}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow 4 \mathrm{CO}_2(\mathrm{~g})+5 \mathrm{H}_2 \mathrm{O}(\mathrm{l}); \quad \Delta_{\mathrm{c}} \mathrm{H}=-2658 \mathrm{~kJ}\)
If a family’s energy requirement per day for cooking is 12000 kJ, how long would a cylinder last?
Solution:
Given
A cylinder of cooking gas contains 11 kg of butane. The thermochemical equation for the combustion of butane is \(\mathrm{C}_4 \mathrm{H}_{10}(\mathrm{~g})+\frac{13}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow 4 \mathrm{CO}_2(\mathrm{~g})+5 \mathrm{H}_2 \mathrm{O}(\mathrm{l}); \quad \Delta_{\mathrm{c}} \mathrm{H}=-2658 \mathrm{~kJ}\)
If a family’s energy requirement per day for cooking is 12000 kJ
Molar mass of butane = 58 g.
58 g of butane produces = 2658 of heat.
Daily requirement of energy = 12000 kJ.
∴ a cyclinder would last = \(\frac{2658 \times 11 \times 10^3}{58 \times 12,000}=\) = 42 days.
Example 2. Calculate the heat of combustion of glucose from the following data.
- \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}); \Delta_{\mathrm{r}} \mathrm{H}_1^{\ominus}=-395.0 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
- \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l}) ; \Delta_{\mathrm{r}} \mathrm{H}_2^{\ominus}=-269.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
- \(6 \mathrm{C}(\mathrm{s})+6 \mathrm{H}_2(\mathrm{~g})+3 \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{C}_6 \mathrm{H}_{12} \mathrm{O}_6(\mathrm{~s}) ; \Delta_{\mathrm{r}} H_3^{\ominus}=-1169.8 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Solution: The required equation for the heat of combustion of glucose is
∴ \(\mathrm{C}_6 \mathrm{H}_{12} \mathrm{O}_6+6 \mathrm{O}_2 \longrightarrow 6 \mathrm{CO}_2+6 \mathrm{H}_2 \mathrm{O} \quad \Delta_{\mathrm{r}} \mathrm{H}^{\ominus}=?\)
Applying Hess’s law, the required equation can be obtained by multiplying (1) by 6 and (2) by 6, adding the products and subtracting (3) from the sum.
∴ \(\begin{aligned}
& 6 \mathrm{C}(\mathrm{s})+6 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 6 \mathrm{CO}_2(\mathrm{~g}) \\
& \frac{6 \mathrm{H}_2(\mathrm{~g})+3\mathrm{O}_2(\mathrm{~g})}{} \longrightarrow 6 \mathrm{H}_2 \mathrm{O}(\mathrm{l}) \\
& \hline 6 \mathrm{H}_2(\mathrm{~g})+6 \mathrm{C}(\mathrm{s})+9 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 6 \mathrm{CO}_2(\mathrm{~g})+6 \mathrm{H}_2 \mathrm{O}(\mathrm{l}) \\
&-\left[6 \mathrm{H}_2(\mathrm{~g})+6 \mathrm{C}(\mathrm{s})+3 \mathrm{O}_2(\mathrm{~g})\right.\left.\longrightarrow \mathrm{C}_6 \mathrm{H}_{12} \mathrm{O}_6(\mathrm{~s})\right] \\
& \hline \mathrm{C}_6 \mathrm{H}_1 \mathrm{O}_6(\mathrm{~s})+6 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 6 \mathrm{CO}_2(\mathrm{~g})+6 \mathrm{H}_2 \mathrm{O}(\mathrm{l})
\end{aligned}\)
Then \(\Delta_{\mathrm{r}} H^{\ominus}\) = \(\Delta_{\mathrm{r}} H^{\ominus}=6 \Delta_{\mathrm{r}} H_1^{\ominus}+6 \Delta_{\mathrm{r}} H_2^{\ominus}-\Delta_{\mathrm{r}} H_3^{\ominus}\)
= 6(-395) + 6(-269.4)-(-1169.8)=-2816.6 kJ.
Enthalpy of neutralisation: The enthalpy change associated with the neutralisation of one gram equivalent of an acid by a base (or vice versa) in a dilute aqueous solution is called the enthalpy of neutralisation. For instance, the enthalpy of neutralisation of NaOH by HCl or HCl by NaOH is -57.1 kJ mol-1. In other words, the enthalpy change associated with the neutralisation of one gram equivalent of HCl by NaOH (or vice versa) is -57.1 kJ mol-1.
∴ \(\mathrm{HCl}(\mathrm{aq})+\mathrm{NaOH}(\mathrm{aq}) \longrightarrow \mathrm{NaCl}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O} ; \quad \Delta_{\mathrm{n}} H^{\ominus}=-57.1 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
The enthalpy of neutralisation of any strong acid by a strong base (or vice versa) is always -57.1 kJ mol-1. This is because neutralisation is actually a reaction between the H+ ions produced by an acid and the OH– ions produced by a base. Let us see how this is so in the case of HCl and NaOH.
∴ \(\mathrm{Na}^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})+\mathrm{H}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l})+\mathrm{Na}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
or \(\mathrm{H}^{+}+\mathrm{OH}^{-} \longrightarrow \mathrm{H}_2 \mathrm{O}\)
- When 1 mol of OH– ions combines with 1 mol of H+ ions to produce 1 mol of water, the heat liberated is 57.1 kJ mol-1. Strong acids and bases are completely ionised in dilute aqueous solutions and the amount of H+ ions or OH– ions produced by one gram equivalent of an acid or a base is always the same, i.e., 1 mol.
- If either the acid or the base, or both, are weak, the enthalpy of neutralisation is usually less than 57.1 kJ mol-1 in magnitude. This is because weak acids and weak bases do not dissociate completely in an aqueous solution and a part of tire energy liberated during the combination of H+ ions and OH– ions is utilised to ionise the weak acid or weak base.
- The heat used for ionising the weak acid (or base) is called the heat of ionisation or dissociation and the net heat of neutralisation is the difference between 57.1 kJ mol-1 and the heat of dissociation.
∴ \(\underset{\text { Weak acdd }}{\mathrm{CH}_3 \mathrm{COOH}}+\underset{\text { Strong base }}{\mathrm{NaOH}} leftrightharpoons \mathrm{CH}_3 \mathrm{COONa}+\mathrm{H}_2 \mathrm{O} ; \quad \Delta \mathrm{H}=-55.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
To calculate the enthalpy of ionisation, let us write down the ionisation reaction and the neutralisation reaction separately.
Ionisation \(\mathrm{CH}_3 \mathrm{COOH} \longrightarrow \mathrm{CH}_3 \mathrm{COO}^{-}+\mathrm{H}^{+} ; \quad \Delta_{\text {ion }} H^{\ominus}=?\)
Neutralisation \(\mathrm{H}^{+}+\mathrm{OH}^{-} \longrightarrow \mathrm{H}_2 \mathrm{O} ; \quad \Delta_{\mathrm{n}} H^{\ominus}=-57.1 \mathrm{~kJ}\)
On adding the two equations, we get, \(\mathrm{CH}_3 \mathrm{COOH}+\mathrm{OH}^{-} \longrightarrow \mathrm{CH}_3 \mathrm{COO}^{-}+\mathrm{H}_2 \mathrm{O}; \Delta_{\text {net }} H^{\ominus}=-55.4 \mathrm{~kJ}\)
The net result is that \(\quad \Delta_{\text {net }} H^{\ominus}=\Delta_{\text {ion }} H^{\ominus}+\Delta_{\mathrm{n}} H^{\ominus} \text { (strong acid-strong base) }\)
or \(\quad-55.4=\Delta_{\text {ion }} H^{\ominus}-57.1 \)
∴\(\quad \Delta_{\text {ion }} H^{\ominus}=-55.4+57.1=2.1 \mathrm{~kJ}\)
In general \(\Delta_{\text {ion }} H^{\ominus}=\left(\Delta_{\text {net }} H^{\ominus}+57.1\right) \mathrm{kJ}\) .
Example: What would be the enthalpy change when
- 0.25 mol of HCl in solution is neutralised by 0.25 mol of a NaOH solution?
- 0.5 mol of nitric acid in solution is mixed with a solution containing 0.2 mol of a potassium hydroxide solution?
- 200 cm3 of a 0.2-M HCl solution is mixed with 300 cm3 of a 0.1-M NaOH solution?
Solution:
1. 0.25 mol of HC1 s 0.25 mol of H+.
0. 25 mol of NaOH = 0.25 mol of OH–.
⇒ \(\mathrm{H}^{+}+\mathrm{OH}^{-} \longrightarrow \mathrm{H}_2 \mathrm{O} \quad \Delta_n H^{\ominus}=-57.1 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Enthalpy change during the formation of 1 mol of H2O = 57.1 kJ.
∴ enthalpy change when 0.25 mol of water is formed = 57.1 x 0.25 = 14.27 kJ.
2. 0.5 mol of HNO3 = 0.5 mol of H+
0. 2 mol of KOH = 0.2 mol of OH–
0. 5 mol of H+ ions will react with 0.2 mol of OH– ions to produce 0.2 mol of H2O.
Enthalpy change = 0.2 x 57.1 = 1142 kj.
3. 200 cm3 of 0.2-M HCl = 0.04 mol of H+.
300 cm3 of 0.1-M NaOH = 0.03 mol of OH–.
0.03 moles of OH– will react with 0.04 mol of H+ to produce 0.03 mol of H2O.
Enthalpy change = 57.1 x 0.03 = 1.71 kj.
Enthalpies of phase change: A phase of a system is a homogeneous part of it throughout which all physical and chemical properties are the same. You will study phases and phase changes in detail later. A couple of examples will give you a basic idea.
- If you have a beaker full of water, the system you are considering consists only of liquid water, so it has one phase. If you drop a few cubes of ice in the water, your system now contains water and ice, so it is a two-phase system.
- When a system changes from one phase to another the process is referred to as a phase change or phase transition. Thus, the conversion of a solid into a liquid (fusion) is a phase change, as also the conversion of a liquid into a gas (vaporisation).
- There are other kinds of phase changes, but we are not concerned with those here. You already know that energy is needed to change a solid to a liquid and a liquid to a gas. The enthalpy changes associated with phase transitions go by different names. Remember, during a phase change, the temperature remains constant.
Enthalpy of fusion The enthalpy of fusion of a substance is the enthalpy change accompanying the conversion of 1 mol of the substance in its solid state into its liquid state at its melting point. For example, the standard enthalpy of fusion of ice is 6.0 kJ mol-1.
∴ \(\mathrm{H}_2 \mathrm{O}(\mathrm{s}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l}) ; \quad \Delta_{\text {fus }} H^{\ominus}=+6.0 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
∴ \(\Delta_{\text {fus }} H^{\ominus}\) is the enthalpy of fusion in the standard state. Freezing is the reverse process of fusion, in that case an equal amount of heat is given off to the surroundings.
∴ \(\Delta_{\text {freex }} H^{\ominus}=-\Delta_{\text {fus }} H^{\ominus}\)
Enthalpy of vaporisation The enthalpy of vaporisation of a substance is the enthalpy change accompanying the conversion of 1 mol of the substance in its liquid state into its vapour state at the boiling point of the liquid. The standard enthalpy of vaporisation of water is 40.7 kj mol-1.
∴ \(\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g}) ; \quad \Delta_{\text {vap }} H^{\ominus}=40.7 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Enthalpy of sublimation The enthalpy of sublimation of a substance is the heat change associated with the conversion of 1 mol of it directly from its solid to its gaseous state at a temperature below its melting point. For instance, the standard enthalpy of sublimation of iodine is 62.39 kJ mol-1.
“Thermodynamics, heat transfer equations, and fundamental principles”
∴ \(\mathrm{I}_2(\mathrm{~s}) \longrightarrow \mathrm{I}_2(\mathrm{~g}) ; \quad \Delta_{\mathrm{sub}} H^{\ominus}=61.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
The enthalpy change of n reverse transition is the negative of the enthalpy change of the forward transition (under the same conditions). This further proves enthalpy to be a state property. Therefore, the change in enthalpy would be same in both the cases when a solid is directly or indirectly converted to vapour. Thus, the enthalpy of sublimation can be expressed as:
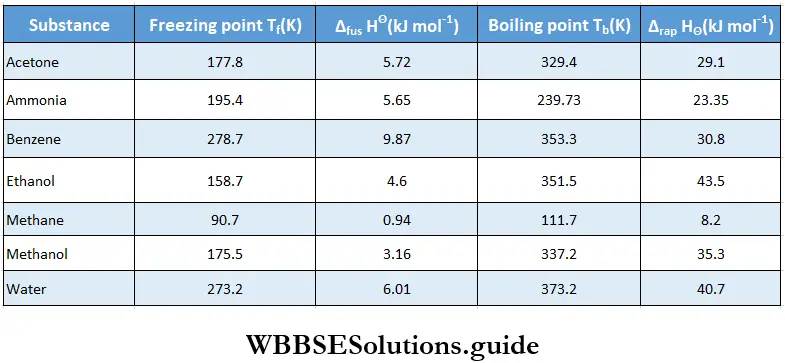
⇒ \(\Delta_{\text {sub }} H^{\ominus}=\Delta_{\text {fus }} H^\ominus+\Delta_{\text {vap }} H^{\ominus}\)
- The striking differences in the magnitude of the enthalpy change for various substances is attributed to the intermolecular interaction in the substances. The enthalpy of vaporisation of water at its boiling point is 40.79 kJ mol-1.
- This signifies that water molecules are held together more tightly than any other liquid with low enthalpy of vaporisation, for instance, acetone. The high enthalpy of vaporisation of water is partly responsible for low humidity in the atmosphere.
Enthalpy of atomisation: The enthalpy change on breaking a molecule completely into its gaseous atoms is called enthalpy of atomisation \(\Delta_{\mathrm{a}} H\). In case of a metal, the enthalpy of atomisation is tine same as the enthalpy of sublimation.
⇒ \(\mathrm{C}(\mathrm{s}) \longrightarrow \mathrm{C}(\mathrm{g}) \Delta_{\mathrm{a}} H^{\ominus}=716.7 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
⇒ \(\mathrm{Na}(\mathrm{s}) \longrightarrow \mathrm{Na}(\mathrm{g}) \Delta_{\mathrm{a}} H^{\ominus}=108.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
The enthalpy of atomisation of diatomic gases is half the bond dissociation enthalpy. For example, the standard bond dissociation enthalpy of O2 is 497 kJ mol-1 and the standard enthalpy of atomisation pertaining to the reaction is 249.2 kJ mol-1.
⇒ \(\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{O}(\mathrm{g})\)
Enthalpy of allotropic transformation: You know that elements like carbon, sulfur, and phosphorus can exist in different allotropic forms and that such elements can change from one allotropic form into another. Allotropic transformations involve enthalpy changes.
- The enthalpy of the allotropic transformation of one allotropic form of a substance into another is the heat change accompanying the transformation per mole of the substance. The standard enthalpy of transformation of rhombic sulphur to monoclinic sulfur, for example, is 1.3 kJ mol-1.
- It is not easy to determine enthalpy changes for allotropic transformations experimentally because such processes are rather slow and the enthalpy changes associated with them are small. Hess’s law can be used conveniently to determine the enthalpy changes accompanying allotropic transformations.
Example: Calculate the enthalpy of the allotropic transformation of rhombic sulphur to monoclinic sulphur from the following thermochemical equations.
- \(
\mathrm{S}\left(\text { rhombic) }+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{SO}_2(\mathrm{~g})\right. \Delta_{\mathrm{c}} H_1^{\ominus}=-295.1 \mathrm{~kJ} \mathrm{~mol}^{-1}\) …(1) - \(\mathrm{~S}(\text { monoclinic })+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{SO}_2(\mathrm{~g}) \Delta_{\mathrm{c}} H_2^{\ominus}=-296.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)……(2)
Solution:
The required equation is S(rhombic) → S(monoclinic) ΔH =?
Applying Hess’s law and subtracting equation (2) from (1)
∴ \(\begin{array}{rc}
\mathrm{S}(\mathrm{r})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{SO}_2(\mathrm{~g}) & \Delta_{\mathrm{c}} H_1=-295.1 \mathrm{~kJ} \\
\mathrm{~S}(\mathrm{~m}) \pm \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{SO}_2(\mathrm{~g}) & \Delta_{\mathrm{c}} H_2=-296.4 \mathrm{~kJ} \\
\hline \mathrm{S}(\mathrm{r}) \longrightarrow \mathrm{S}(\mathrm{m}) & \Delta H=13 \mathrm{~kJ}
\end{array}\)
Therefore, the enthalpy of the allotropic transformation of rhombic sulphur to monoclinic sulphur is 1.3 kJ mol-1.
Enthalpy of solution: The enthalpy of reaction for dissolving one mole of a solute in n moles of a solvent is known as the integral enthalpy of the solution or the integral heat of the solution. Let us look at some such values for a solution of HNO3 in water at 298 K.
1. \(\left.\mathrm{HNO}_3(\mathrm{l})+\mathrm{H}_2 \mathrm{O} \longrightarrow \mathrm{HNO}_3 \text { (in } 1 \mathrm{H}_2 \mathrm{O}\right) \Delta_{\text {sol }} H^{\ominus}=-187.6 \mathrm{~kJ} \mathrm{~mol}^{-1}\) ….(1)
2. \(\left.\mathrm{HNO}_3(\mathrm{l})+10 \mathrm{H}_2 \mathrm{O} \longrightarrow \mathrm{HNO}_3 \text { (in } 10 \mathrm{H}_2 \mathrm{O}\right) \Delta_{\text {sol }} H^{\ominus}=-205.8 \mathrm{~kJ} \mathrm{~mol}^{-1}\) …. (2)
3. \(\mathrm{HNO}_3(\mathrm{l})+100 \mathrm{H}_2 \mathrm{O} \longrightarrow \mathrm{HNO}_3 \text { (in } 100 \mathrm{H}_2 \mathrm{O} \text { ) } \Delta_{\text {sol }} H^{\ominus}=-206.8 \mathrm{~kJ} \mathrm{~mol}^{-1}\)…. (3)
4. \(\mathrm{HNO}_3(\mathrm{l})+500 \mathrm{H}_2 \mathrm{O} \longrightarrow \mathrm{HNO}_3 \text { (in } 500 \mathrm{H}_2 \mathrm{O} \text { ) } \Delta_{\text {sol }} H^{\ominus}=-206.9 \mathrm{~kJ} \mathrm{~mol}^{-1}\)…..(4)
5. \(\left.\mathrm{HNO}_3(\mathrm{l})+1000 \mathrm{H}_2 \mathrm{O} \longrightarrow \mathrm{HNO}_3 \text { (in } 1000 \mathrm{H}_2 \mathrm{O}\right) \Delta_{\text {sol }} H^{\ominus}=-206.9 \mathrm{~kJ} \mathrm{~mol}^{-1}\)….(5)
6. \(\mathrm{HNO}_3(\mathrm{l})+\mathrm{aq} \longrightarrow \mathrm{HNO}_3 \text { (aq) } \Delta_{\text {sol }} H^{\ominus}=-207.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\) …..(6)
As you can see from Equations (1)-(6), the heat of solution varies with concentration or decreases with increase in the amount of the solvent and finally stabilises at infinite dilution.
The enthalpy of solution at infinite dilution is the enthalpy change when 1 mol of the substance dissolves in such a large amount of solvent that the interaction between the solute molecules is negligible. In case of water as a solvent, infinite dilution is denoted by ‘aq’ (aqueous).
For HNO3, the enthalpy of solution at infinite dilution refers to Equation (6) and is -207.36 kJ mol-1.
Some more values of enthalpy of solution at infinite dilution are given below.
⇒ \(\mathrm{KCl}(\mathrm{s})+\mathrm{aq} \longrightarrow \mathrm{KCl}(\mathrm{aq}) ; \Delta_{\mathrm{sol}} H^{\ominus}=+18.6 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
⇒ \(\mathrm{CuSO}_4(\mathrm{~s})+\mathrm{aq} \longrightarrow \mathrm{CuSO}_4(\mathrm{aq}) ; \Delta_{\mathrm{sol}} H^{\ominus}=-66.5 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
⇒ \(\mathrm{CuSO}_4 \cdot 5 \mathrm{H}_2 \mathrm{O}+\mathrm{aq} \longrightarrow \mathrm{CuSO}_4(\mathrm{aq}) ; \Delta_{\text {sol}} H^{\ominus}=+11.7 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
- In the first and third cases \(\Delta_{\text {sol}}\) is positive (endothermic), while in the second, it is negative (exothermic). In general, salts which do not form hydrates dissolve in water with the absorption of heat. Hydrated salts also dissolve in water with the absorption of heat. The process is exothermic only in the case of anhydrous salts which form hydrates.
- When a substance dissolves in water, it either absorbs (positive enthalpy) or releases energy (negative enthalpy). Ammonium nitrate has a positive enthalpy of solution. Tire cold pack used for minor injuries to sportsmen contains NH4NO3 and water in separate compartments.
- It is activated on punching the partition between the two upon which the salt dissolves and the temperature of water falls. Calcium chloride, on the other hand, has a negative enthalpy of solution, and this phenomenon is used to dissolve ice on sidewalks in winters.
- Let us see what happens when NaCl dissolves in water. First, the three-dimensional network of Na+ and Cl– breaks into individual ions. For this purpose the lattice energy which is responsible for holding the ions together must be overcome. The separated Na+ and Cl– ions get stabilised in tire solution by their interaction with the water molecules.
This process involves breaking some of the hydrogen bonds between the water molecules. The ions are then said to be hydrated. The process of dissolution of an ionic compound in a solvent (water) involves a complex interaction between the solute and the solvent species. However, it can be broadly taken to be happening in two steps:
1. First the ionic lattice breaks: \(\mathrm{NaCl}(\mathrm{s}) \longrightarrow \mathrm{Na}^{+}(\mathrm{g})+\mathrm{Cl}^{-}(\mathrm{g})\)
Energy is required for this process and therefore the reaction is endothermic. The enthalpy change associated with it is called lattice enthalpy it may be defined as the heat change when one mole of an ionic solid separates into its constituent gaseous atoms.
2. Next the gaseous Na+ and Cl– ions interact with water molecules and become hydrated:
⇒ \(\mathrm{Na}^{+}(\mathrm{g})+\mathrm{Cl}^{-}(\mathrm{g}) \stackrel{\mathrm{H}_2 \mathrm{O}}{\longrightarrow} \mathrm{Na}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
The enthalpy change associated with this process is called the enthalpy of solvation or more precisely the enthalpy of hydration as water is the solvent.
∴ \(\Delta_{\text {sol }} H=\Delta_{\text {lattlee }} H+\Delta_{\text {hyd }} H\)
Lattice enthalpy: It is not possible to determine lattice energy directly by experiments. Hence, it is determined on the basis of the Born-Haber cycle. The Bom-Haber cycle is a closed sequence of different processes which are involved in the breaking and finally in the making of an ionic crystal. To understand the cycle, let us take the example of NaCl(s). The steps involved in the formation of NaCl(s) are as follows:
1. Sublimation of Na(s) to Na(g): \(\mathrm{Na}(\mathrm{s}) \longrightarrow \mathrm{Na}(\mathrm{g}) \quad \Delta H=\Delta_{\mathrm{a}} H^{\ominus}[\mathrm{Na}(\mathrm{s})]=108.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
2. Dissociation of Cl2(g) to give Cl(g): \(\frac{1}{2} \mathrm{Cl}_2(\mathrm{~g}) \longrightarrow \mathrm{Cl}(\mathrm{g}) \quad \Delta H=\frac{1}{2} \Delta_{\mathrm{Cl}-\mathrm{C}} H^{\ominus}=\frac{1}{2} \times 242 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
3. Ionisation of Na(g): \(\mathrm{Na}(\mathrm{g}) \longrightarrow \mathrm{Na}^{+}(\mathrm{g})+\mathrm{e}^{-} \quad \Delta H=\Delta_{\text {ien }} H^{\ominus}=495.8 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Ionisation enthalpy of sodium = 495.8 kJ mol-1. Since sodium is in the gaseous state, the enthalpy change is given by
∴ \(\Delta_{\text {ion }} H=\Delta_{\text {ion }} U+\Delta n_g R T\)
∴ \(\Delta_{\text {ion }} H=495.8+(1) \times R T\)
(It may be observed subsequently that RT cancels out with -RT in the next step.)
4. The electron released by the sodium atom is gained by chlorine to give Cl–.
⇒ \(\mathrm{Cl}(\mathrm{g})+\mathrm{e}^{-} \longrightarrow \mathrm{Cl}^{-}(\mathrm{g})\)
The electron affinity (EA) of Cl is 348.6 kJ mol-1. Since electron affinity is defined as the energy released in the above process, from thermodynamic considerations we must take a negative value.
Hence energy involved (-EA) =-348.6 kJ mol-1. Here again, we need the electron gain enthalpy (\(\Delta_{\mathrm{eg}} H)\), which is -EA – RT (since Δng = -1)
∴ \(\Delta_{\mathrm{eg}} H)\) = -348.6 + (-1)RT = -348.6 – RT.
5. Na+(g) and Cl– (g) (steps 3 and 4) combine to give NaCl(s).
⇒ \(\mathrm{Na}^{+}(\mathrm{g})+\mathrm{Cl}^{-}(\mathrm{g}) \longrightarrow \mathrm{NaCl}(\mathrm{s})\)
Enthalpy change for this process = –\(\Delta_{\text {Laltice }} H\)(The process involves the formation of the lattice.) These steps are depicted as the sequence of steps in the Bom-Haber cycle.

The basis of calculations using the above cycle is that enthalpy being a state function, the sum of enthalpy changes around the cycle (clockwise direction) is zero.
∴ \(-\Delta_{\mathrm{f}} H[\mathrm{NaCl}(\mathrm{s})]+\Delta_{\text {sub }} H[\mathrm{Na}(\mathrm{s})]\)+\(\frac{1}{2} \Delta_{\text {diss }} H\left[\mathrm{Cl}_2(\mathrm{~g})\right]+I \cdot E .+R T-E . A-R T-\Delta_{\text {lattice }}\) H =0
or \(\Delta_{\text {lattice }} H=-\Delta_{\mathrm{f}} H+\Delta_{\text {sub }} H+\frac{1}{2} \Delta_{\text {diss }} H+I \cdot E .-E . A .\)
The \(\Delta_{\mathrm{f}}H\) of NaCl(s) is -411.2 kJ.
Substituting the appropriate values on the RHS, we get
∴ \(\Delta_{\text {Lattice }} H =411.2+108.4+\frac{1}{2} \times 242+495.8-348.6\) =1136.4-348.6
∴ \(\Delta_{\text {Lattice }} H =787.8 \mathrm{~kJ}\).
Bond energy: By now you know what enthalpies of formation of compounds are. You also know that compounds are formed by the breaking and making of bonds. To have a precise knowledge of the enthalpy change for a process, the bond dissociation enthalpy or the bond dissociation energy [ΔH(A-B)] is considered.
For example, when the dissociation, or breaking, of a chemical bond occurs as in the following process,the corresponding molar enthalpy change is called the bond dissociation enthalpy. Thus, the bond dissociation enthalpy is tire enthalpy associated with the breaking of 1 mol of a substance in the gaseous state completely into its gaseous atoms.
∴ AB(g) → A(g) + B(g)
The values of bond enthalpies depend on the bonding present between two atoms in the molecule. Even in the same molecule, the values may differ. For example, to break the first O—H bond in water, the enthalpy change \(\Delta H^{\ominus}(\mathrm{HO}-\mathrm{H})\)= 492 kJ mol-1 while for breaking the second bond, \(\Delta H^{\ominus}(\mathrm{O}-\mathrm{H})\) is 428 kJ mol-1.
- In methane four hydrogen atoms are attached to a carbon atom and the four C—H bonds are equal in energy and bond length. In this case the total enthalpy is expected to be four times the C—H bond enthalpy.
- However, this is not the case, the reason being different dissociation steps. The first step involves the breaking of a C—H bond in the CH4 molecule but in the next step the C—H bond is broken in a CH3 radical, and so on.
The energies required to break the individual C—H bonds in different dissociation steps are as follows.
∴ \(\mathrm{CH}_4(\mathrm{~g}) \longrightarrow \mathrm{CH}_3(\mathrm{~g})+\mathrm{H}(\mathrm{g}) ; \Delta_{\text {bond }} H^{\ominus}=427 \mathrm{~kJ} \mathrm{~mol}^{-1},\)
∴ \(\mathrm{CH}_3(\mathrm{~g}) \longrightarrow \mathrm{CH}_2(\mathrm{~g})+\mathrm{H}(\mathrm{g}) ; \Delta_{\text {bond }} H^{\ominus}=439 \mathrm{~kJ} \mathrm{~mol}^{-1},\)
∴ \(\mathrm{CH}_2(\mathrm{~g}) \longrightarrow \mathrm{CH}(\mathrm{g})+\mathrm{H}(\mathrm{g}) ; \Delta_{\text {bond }} H^{\ominus}=452 \mathrm{~kJ} \mathrm{~mol}^{-1},\)
∴ \(\mathrm{CH}(\mathrm{g}) \longrightarrow \mathrm{C}(\mathrm{g})+\mathrm{H}(\mathrm{g}) ; \Delta_{\text {bond }} H^{\ominus}=347 \mathrm{~kJ} \mathrm{~mol}^{-1}\).
The total enthalpy change for the atomisation of methane \(\left[\mathrm{CH}_4(\mathrm{~g}) \longrightarrow \mathrm{C}(\mathrm{g})+4 \mathrm{H}(\mathrm{g})\right] \text { is } \Delta_{\mathrm{a}} H^{\ominus}\) = 166 x 103 kJ mol-1.
In case of reactions for which experimental data are not available for successive steps, mean bond enthalpy is used. It is the value of bond dissociation energy of a bond A—B averaged over a series of related compounds. For example, the bond energy for the O—H bond, \(\Delta H^{\ominus}\)(O—H) (averaged between H2O and alcohols) is 463 kJ mol-1.
Similarly, in case of CH4, all the bond enthalpies added together come to 1665 kJ mol-1. The mean bond enthalpy of C—H bond in CH4 is
⇒ \(\frac{1}{4} \Delta_a H^{\ominus}=\frac{1}{4} \times 1665=416 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Now consider the atomisation of a diatomic molecule: \(\mathrm{H}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{H}(\mathrm{g}) ; \quad \Delta_a H^{\ominus}=435 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Since two atoms are formed, the energy value is the enthalpy of atomisation of H2 at 298 K.
The corresponding bond energy \(\Delta_a U^{\ominus}\) for the same reaction (with a little difference from the enthalpy) is 430.8 kJ mol-1. That is why the two quantities are often used interchangeably. But the correct way would be to convert from ΔU to ΔH.
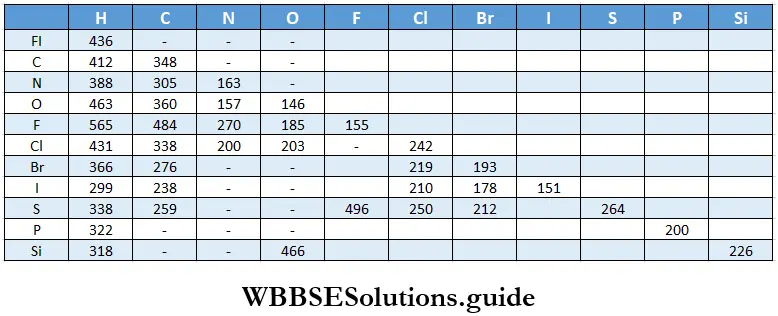
We know that ΔH = ΔU + pΔV
Some important bond enthalpies (kJ mol-1) of multiple bonds are as follows.
C—C = 612 N=N = 418 O=O= 497
C≡C = 837 N≡N = 946
C=O = 741 C=N = 615
- For gases, pΔV (at constant pressure) may be replaced by RT. In case of a diatomic molecule, the bond energy is equal to the bond dissociation energy.
- The dissociation energy may here be defined as the enthalpy change involved in breaking the bond between atoms of a gaseous diatomic molecule. Also, it can be seen that bond enthalpy in homonuclear diatomic molecules is twice the enthalpy of formation for the atom in the gaseous state.
Bond energies can be used to calculate the enthalpy of a reaction. The standard reaction enthalpy, \(\Delta_r H^{\ominus}\) is the difference between the sum of the standard enthalpies of the reactants and products.
∴ \(\Delta_r H^{\ominus}=\Sigma \text { bond enthalpies }{ }_{\text {reactants }}-\Sigma \text { bond enthalpies products }\)
This relationship is valid when all the substances in a chemical reaction are in the gaseous state.
Example 1. Calculate the enthalpy change for the following reaction. \(2 \mathrm{C}_2 \mathrm{H}_2(\mathrm{~g})+5 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 4 \mathrm{CO}_2(\mathrm{~g})+2 \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
The average bond energies of the C—H, C≡C, O=O, C—O, and O—H bonds are 414, Hit), 499, 724, and 460 kJ mol-1 respectively.
Solution:
ΔH = sum of bond energies of reactants – sum of bond energies of products.
The given reaction can be written as 2H—C≡C—H(g) + 5O=O(g) → 4O=C=)(g) + 2H—O—H(g)
∴ \(\Delta H=\left[2 \Delta H_{\mathrm{C} \equiv \mathrm{C}}+4 \Delta H_{\mathrm{C}-\mathrm{H}}+5 \Delta H_{\mathrm{O}=\mathrm{O}}\right]-\left[8 \Delta H_{\mathrm{C}=\mathrm{O}}+4 \Delta H_{O-H}\right]\)
= (2 x 810 + 4 x 414 + 5 x 499) – (8 x 724 + 4 x 460)
= 5771 – 7632 = -1861 kJ mol-1.
Example 2. Calculate the bond energy of the C—H bond in CH4 front the following data.
- \(\mathrm{C}(\mathrm{s})+2 \mathrm{H}_2(\mathrm{~g}) \longrightarrow \mathrm{CH}_4(\mathrm{~g}) \quad \Delta H_1^{\ominus}=-74.8 \mathrm{~kJ} \mathrm{~mol}^{-1}\) ….(1)
- \(\mathrm{H}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{H}(\mathrm{g}) \quad \Delta \mathrm{H}_2^{\ominus}=435.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\) …..(2)
- \(\mathrm{C}(\mathrm{s}) \longrightarrow \mathrm{C}(\mathrm{g}) \quad \Delta H_3^{\ominus}=718.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)….(3)
Solution:
To find the bond energy of the C—H bond in CH4, let us first write the equation for the dissociation of CH4 into C and H, The enthalpy change for this reaction would be four times the bond energy of the C—H bond since the dissociation of the CH4 molecule involves the breaking of four C—H bonds.
CH4 (g) → C(g) + 4H(g) ΔH = ?
The enthalpy change for this reaction can be calculated from the given data by applying Hess’s law. The equation for this reaction can be obtained by multiplying Equation (2) by 2, adding the product to Equation (3), and then subtracting Equation (1) from the sum.
∴ \(\begin{aligned}
\mathrm{C}(\mathrm{s})+2 \mathrm{H}_2(\mathrm{~g}) & \longrightarrow 4 \mathrm{H}(\mathrm{g})+\mathrm{C}(\mathrm{g}) \\
\mathrm{C}(\mathrm{s})+2 \mathrm{H}_2(\mathrm{~g}) & \longrightarrow \mathrm{CH}_4(\mathrm{~g}) \\
\hline \mathrm{CH}_4(\mathrm{~g}) & \longrightarrow 4 \mathrm{H}(\mathrm{g})+\mathrm{C}(\mathrm{g})
\end{aligned}\)
∴ ΔH = 2 x ΔH2 + ΔH3 – ΔH1
= 2 x 435.4 + 718.4-(-748)
= 870.8 + 718.4 + 748 = 16640 kJ mol-1.
Thus, the energy required to break four C—H bonds = 1664.0 kJ mol-1.
Therefore, the bond energy of the C—H bond = 1664/4 = 416 kJ mol-1.
Alternatively, the enthalpy of the formation of the reactants and products can also be used to find the enthalpy change for the required reaction.
ΔH = ∑ΔHf (products) -∑ΔHf (reactants)
ΔH = [ΔH3 +2ΔH2]-ΔH1
= [718.4 + 2 x 435.4] – [-748]
= 718.4 + 870.4 + 74.8 = 1664.0 kJ mol-1,
∴bond energy of C—H bond = 1664/4 = 416 kJ mol-1.
The Measurement Of Heat
In the laboratory, heat changes in physical and chemical processes are measured with a calorimeter. Calorimetry is the measurement of heat changes associated with a process. The study of calorimetry involves the concept of specific heat and heat capacity. So let us define these first
The heal capacity (C) of a substance is the amount of heat required to raise the temperature of a given quantity of it by 1°C Heat capacity is an extensive property. It is more convenient to use an intensive property. Therefore, the molar heat capacity, Cm (Cm = C/n in JK-1 mol-1 where n is the amount of the substance), is used. The specific heat capacity Cs (Cs = C/m in JK-1 g-1, where m is the mass of the substance) is also an intensive property.
For water, the specific heat is \(1 \mathrm{cal} \mathrm{g}^{-1} \mathrm{C}^{-1} \text { or } 4.18 \mathrm{~J} \mathrm{~g}^{-10} \mathrm{C}^{-1}\)
The molar heat capacity of a substance is the heat required to raise the temperature of 1 mol of the substance by one degree.
The heat capacity at constant volume (Cv) is given by \(C_v=\frac{q_v}{\Delta T} .\)
From the first law of thermodynamics, we have q = ΔU ÷ pΔV.
At constant volume, ΔV = 0. qv = AU.
∴ heat capacity at constant volume is given by \(C_V=\left(\frac{\Delta U}{\Delta T}\right)_V\)
Thus, the heat capacity at constant volume is defined as the rate of change of internal energy with temperature.
The heat capacity at constant pressure is given by \(C_p=\frac{q_p}{\Delta T}.\)
We have already learned that, at constant pressure, ΔH = qp.
Therefore, heat capacity at constant pressure is given by \(C_p=\left(\frac{\Delta H}{\Delta T}\right)_p\)
Thus, the heat capacity at constant pressure is defined as the rate of change of enthalpy with temperature. Relationship between Cp and Cv We know that, at constant pressure (Equation 6),
ΔH = ΔU + pΔV.
For 1 mole of an ideal gas, pΔV = RΔT (ideal gas equation).
Thus, Equation 6 can be written as ΔH = ΔU + RΔT
On dividing the equation by ΔT, we get \(\frac{\Delta H}{\Delta T}=\frac{\Delta U}{\Delta T}+R\)
or Cp = Cv + R
or Cp – Cv = R
Cp is always greater than Cv for any gas, since in a constant-pressure process a system has to do work against the surroundings.
Coming back to the measurement of energy changes associated with chemical or physical processes, measurements are made under two different conditions:
- at constant volume, ΔU -qv, and
- at constant pressure, ΔH = qp.
Constant-volume calorimetry: The reactions that can be most easily studied under constant- volume conditions are combustion reactions. The apparatus used for this purpose is called the bomb calorimeter or adiabatic bomb calorimeter. It consists of a sealed constant-volume steel container (known as a bomb) in which the reaction occurs. The bomb can withstand high pressures.
- A known mass of a combustible substance is placed in the bomb, which is filled with oxygen at 30 atm pressure. The sealed bomb is then immersed in a known amount of water contained in an insulated container. The whole set-up is called the calorimeter. Since the outer covering is insulated, there is no heat exchange with the surroundings and hence the reaction process is adiabatic.
- The sample is ignited electrically and the heat produced by the combustion reaction is absorbed by the water surrounding the bomb and the bomb itself. The temperature of the water is monitored and the heat produced is calculated using the equation
∴ \(q=C_{\text {cal }} \Delta T,\) ….(1)
where Ccal is the heat capacity of the calorimeter. Ccal is determined separately by igniting a known amount of a substance whose heat of combustion is already known, in the same calorimeter.
- Thus the heat produced in the reaction may be determined. We already know that at constant volume qv = ΔU. In constant- volume calorimetry, the heat change observed is simply the change in internal energy.
- This value may be modified to get the enthalpy, but the pressure changes are usually small and may be neglected. The heat changes may be simply assumed to correspond to enthalpy changes.
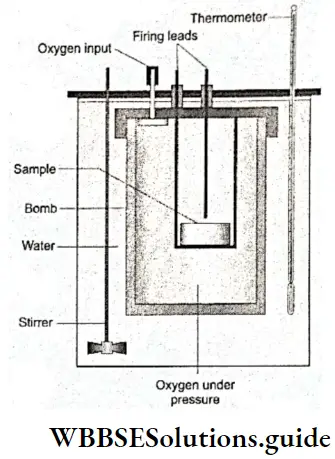
Constant-pressure calorimetry: To measure the heat changes at constant pressure, a device comparable to a thermos flask is used. The calorimeter is just a vessel with a stirrer and a thermometer.
The system is insulated from the surroundings and hence acts as an isolated system. The heat changes within the vessel are measured in terms of the change in temperature of the system. Constant monitoring of the temperature before and after the reaction helps in finding q and hence AH, as we know that, ΔH = qp
The amount of heat exchanged (qv or qp) in any process is given by, qp =msΔT……(2)
where m is the mass of the substance, s is its specific heat and ΔT is the change in the temperature.
You already know that q = CΔT =Cp ΔT (at constant pressure),
where C = ms, the heat capacity of the system.
- Thus to determine q, C and ΔT have to be known first. The experimental technique—calorimetry—actually involves two steps, the first being the determination of the heat capacity of the calorimeter.
- The second step involves the determination of the change in temperature during the completion of the reaction. Since it is an insulated system, no heat is lost to the surroundings as in the case of constant-volume calorimetry.
As you already know, the enthalpy of the reaction is simply the heat change at constant pressure, ΔH = qp (at constant pressure).
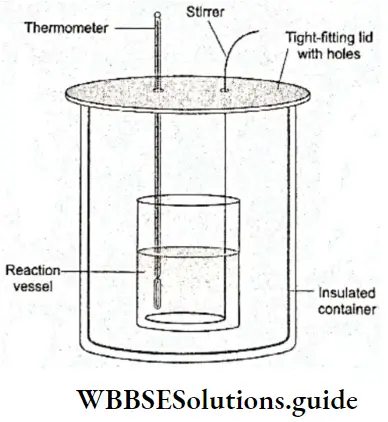
Enthalpy changes are commonly tabulated at 25°C and it is often required to know their values at other temperatures. They can be calculated from the heat capacities of the reactants and products.
You know that ΔH = H(products) – H(reactants).
At constant pressure ΔH /ΔT = Cp.
Therefore, for small changes in temperature, \(\frac{\Delta(\Delta H)}{\Delta T}=C_p \text { (products) }-C_p \text { (reactants) }=\Delta C_p \Delta T\)
∴ \(\Delta(\Delta H)=\Delta C_p\left(T_2-T_1\right)\)
or \(\Delta H_2-\Delta H_1=\Delta C_p\left(T_2-T_1\right)\),
where ΔH2 is the enthalpy of the reaction at T2 and ΔH1 is that at T1.
Example 1. Calculate the enthalpy change on the freezing of 1 mol of zoater at -5°C to ice at -5°C. Given that
⇒ \(\Delta_{\text {tom }} H^{\oplus}\left(\mathrm{H}_2 \mathrm{O}\right)=6.03 \mathrm{~kJ} \mathrm{~mol}^{-1} \text { at } 0^{\circ} \mathrm{C} \text { and } C_p\left[\mathrm{H}_2 \mathrm{O}(\mathrm{l})\right]\)
= \(75.3 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1}, C_{,},\left[\mathrm{H}_2 \mathrm{O}(\mathrm{s})\right]=36.8 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} \text {. }\)
Solution:
To find the enthalpy change at -5°C, we use the equation: \(\Delta H_2-\Delta H_1=\Delta C_p\left(T_2-T_1\right) .\)
First, we will find Δfus H at -5°C and then convert it to ΔH (freezing) by prefixing a (-) sign to ΔfusH.
Also, ΔH is in kJ and Cp in J. Converting, ΔfusH = 6.03 kJ mol-1 = 6.03 x 1000 J mol-1.
∴ \(\Delta H_2-6.03 \times 1000= (75.3-36.8)\times(-5-0) \)
= \(C_p(1)-C_p(\mathrm{~s})=-192.5\)
∴ \(\Delta H_2=-192.5+6030=5837.5\)
Thus \(\Delta_{\text {fus }} H at -5^{\circ} \mathrm{C}=5837.5 \mathrm{~J} \mathrm{~mol}^{-1}=5.84 \mathrm{~kJ} \mathrm{~mol}^{-1} and \Delta H (freezing) at -5^{\circ} \mathrm{C}\)
= \(-5.84 \mathrm{~kJ} \mathrm{~mol}^{-1}\).
“Laws of thermodynamics, equations, and real-life applications”
Example 2. Find the heat required to raise the temperature of 27.9 g of Fe from 30 to 40°C. The molar heat capacity of Fe(s) =25.10 J K-1mol-1.
Solution:
The molar heat capacity is the heat required to raise the heat of 1 mol of the substance, i.e., 55.8 g (molar mass of Fe) by 1°C.
The heat required to raise the temperature of 55.8 g of Fe by 1°C = 25.1 J.
The heat required to raise the temperature of 27.9 g of Fe by 1°C = \(\frac{25.1}{55.8}\) x 27.9 J.
∴ the heat required to raise the temperature of 27.9 g of Fe by 10°C = \(\frac{25.1}{55.1}\)x 27.9 x 10 = 125.5 J.
Example 3. 1.922 g of methanol (CH3OH) was burnt in a constant-volume bomb calorimeter. The temperature of water rose by 4.2°C. If the heat capacity of the calorimeter and its contents is 10.4 kJ °C-1, calculate the molar heat of combustion of methanol.
Solution:
Given
1.922 g of methanol (CH3OH) was burnt in a constant-volume bomb calorimeter. The temperature of water rose by 4.2°C. If the heat capacity of the calorimeter and its contents is 10.4 kJ °C-1,
If qcal is the quantity of heat involved in the reaction and C„ is the heat capacity of the calorimeter, then
∴ \(q_{\text {eat }}=C_V \Delta T\)
But \(q_{\text {cal }}=-q_r\)
∴ \(\quad q_r=-C_v \Delta T\)
= \(-10.4 \times 4.2\left[because \text { the temperature rose by } 4.2^{\circ} \mathrm{C}, \Delta T \text { is positive }\right]\) = \(-43.68 \mathrm{~kJ}\).
The minus sign indicates exothermic nature of the reaction.
Since the pressure changes are small and can be neglected, qr = ΔHr.
Tim ΔH for burning 1.922 g of methanol is -43.68 kJ.
The ΔH for burning 1 mol or 32 g of methanol is obtained as
∴ \(\Delta H=\frac{-43.68 \mathrm{~kJ}}{1.922 \mathrm{~g}} \times 32 \mathrm{~g} \mathrm{~mol}^{-1}\) = -727.24 kJ mol-1.
The molar enthalpy or molar heat of combustion is therefore -727.24 kJ mol-1.
Spontaneous Processes
Generally speaking, when we say something has happened spontaneously, we mean that it has happened of its own accord. In chemistry, the meaning is a little different. It would be easy enough for you to associate the word spontaneous with a process like the dissolving of sugar in water, or the evaporation of water from an open vessel.
- But the burning of coal in air, or the burning of ethane to produce carbon dioxide and water are also spontaneous processes, even though these processes have to be initiated by ignition.
- Any process which can occur under a given set of conditions is called a spontaneous process, irrespective of whether it occurs on its own, or needs to be initiated. In other words, a spontaneous process occurs on its own or has a tendency to occur. A simple way of saying this is that a spontaneous process is a process which is possible.
- It is not possible, for example, to dissolve sand in water, so this is not a spontaneous process. Water cannot flow uphill, so this is not a spontaneous process either. Some nonspontaneous processes can, however, be made to occur by providing energy from an external source.
- For instance, water can be made to flow uphill by using a pump. Similarly, though the electrolysis of water is not a spontaneous process, it can be made to occur by supplying electrical energy.
You may wonder about the difference between a nonspontaneous process which can be made to occur by supplying energy and a spontaneous process which requires initiation. The combination of gaseous hydrogen and oxygen to form water is a spontaneous reaction which has to be initiated by an electric spark.
∴ \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
- The electrolysis of water is a nonspontaneous process which requires electrical energy. The difference is that once a spontaneous reaction has been initiated, it proceeds on its own, while a nonspontaneous process stops as soon as the supply of external energy stops.
- Once the electric spark starts the reaction between hydrogen and oxygen, the process continues on its own. But if the supply of electric current is stopped during the electrolysis of water, the process stops.
Criterion for spontaneity: What makes certain processes possible or spontaneous, and certain others not possible or nonspontaneous? What is the driving force behind the occurrence of different processes? If we know this we can predict whether a certain process would be possible.
- You have already read in several contexts that every system seeks to have the minimum possible energy in order to acquire the maximum stability. Water flows down a hill to have the minimum possible potential energy. A wound spring unwinds itself to minimise energy. Atoms form bonds with each other in order to have less energy.
- This may lead you to the conclusion that the criterion for a process is the reduction of energy, or that a process is possible if it causes a reduction in the energy of the system. Let us consider some chemical reactions and see if this is right. In exothermic reactions, the energy of the products is less than the energy of the reactants and the reaction is accompanied by the evolution of heat.
⇒ \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \Delta H =-394 \mathrm{~kJ}\)
⇒ \(\mathrm{~N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{NH}_3(\mathrm{~g}) \Delta H =-92 \mathrm{~kJ}\)
In such cases at least we may be right in saying that the tendency to attain a state of minimum energy (i.e., negative enthalpy change) is the driving force behind the occurrence of a reaction. But what about endothermic reactions which are spontaneous?
⇒ \(\mathrm{H}_2 \mathrm{O}(\mathrm{s}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l}) \Delta H=5.86 \mathrm{~kJ}\)
⇒ \(\mathrm{NH}_4 \mathrm{Cl}(\mathrm{s})+\mathrm{aq} \longrightarrow \mathrm{NH}_4^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq}) \Delta H=177.8 \mathrm{~kJ}\)
⇒ \(\mathrm{CaCO}_3(\mathrm{~s}) \longrightarrow \mathrm{CaO}(\mathrm{s})+\mathrm{CO}_3(\mathrm{~g}) \Delta H=15.1 \mathrm{~kJ}\)
- These three reactions involve the absorption of heat, which means that the reactions lead to an increase in the energy of the system, so reduction of energy cannot be the only criterion determining the spontaneity of a reaction.
- Reversible reactions also prove that decrease in energy or negative enthalpy change cannot be the only determining factor for the feasibility of a reaction. Suppose the forward reaction is exothermic then the backward reaction has to be endothermic and it would not occur if the only criterion for spontaneity were reduction of energy.
The tendency towards maximum disorder: To investigate what other factors could possibly determine the spontaneity of a process, let us consider a process for which ΔH = 0, since for such a process the energy factor could not be the driving force. The mixing of air and bromine vapour is just such a process.
- Suppose vessel A contains bromine vapour and vessel B contains air and there is a movable partition between the two. Now suppose that the partition is removed. The two gases will mix completely. What makes this possible? When the samples of air and bromine vapour are in two separate vessels, there is more order and when they diffuse into each other, there is more disorder.
- This tendency of a system to move towards greater disorder or to maximise randomness is the other driving force which makes reactions possible. It explains why endothermic reactions are possible despite the fact that ΔH is positive for such reactions. Take the case of the dissolution of ammonium chloride in water.
⇒ \(\mathrm{NH}_4 \mathrm{Cl}(\mathrm{s})+\mathrm{aq} \longrightarrow \mathrm{NH}_4^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
- This is an endothermic process, but it is possible because the randomness of the system increases. How? When the ions are held together in the crystal lattice there is more order and when they go into the aqueous solution, they are more free to move about, hence there is more disorder.
- Just as the energy factor (minimising energy) cannot be the only criterion which determines the spontaneity of a process, nor can maximising randomness be the sole determining factor. If this were the case, the liquefaction of a gas (which increases order or decreases randomness) would not have been possible. But we will come to that later.
Entropy: The randomness or disorder of a system is measured in terms of a function called entropy. It is a state function, represented by S and measured in J K-1.
Change in entropy (ΔS) = Sfinal state – Sintial state
or ∑ Sproducts – ∑ Sreatants
When ΔS for a process is positive, the process leads to an increase in randomness or disorder.
In the examples discussed earlier, the processes are accompanied by an increase in entropy. We may again tend to conclude that for spontaneous processes there must be an increase in entropy. However, there are cases where entropy decreases. For example, the spontaneous condensation of steam to liquid water.
⇒ \(\underset{s^{\ominus}=188.7 \mathrm{JK}^{-1} \mathrm{~mol}^{-1}}{\mathrm{H}_2 \mathrm{O}(\mathrm{g})} \longrightarrow \underset{s^{\ominus}=70.0 \mathrm{JK}^{-1} \mathrm{~mol}^{-1}}{\mathrm{H}_2 \mathrm{O}(\mathrm{l})}\)
- The standard molar entropy values for liquid water and water vapour are given and show that there is a decrease in entropy. Standard entropy may be defined in a manner similar to standard enthalpy.
- The standard molar entropy of a substance is the entropy of one mole of the pure substance at 1 bar pressure and a specified temperature, usually 298 K. Let us not be in a hurry to make conclusions. When steam condenses, energy is given out, \(\Delta H^\ominus\) being -44.1 kJ mol-1 for the reaction.
- Where does this energy go? If the water condenses on a window, the energy is passed on to the window and when it condenses in the air, energy is taken up by the air molecules. Therefore, the entropy of the surroundings increases.
- The tendency towards disorder is summed up in the second law of thermodynamics, which states that spontaneous reactions are accompanied by a net increase in the entropy of the universe. Here the universe means the system and surroundings taken together,
⇒ \(\Delta S_{\text {univ }}>0 \) (2nd law thermodynamics).
⇒ \(\Delta S_{\text {uriv }}=\Delta S_{\text {sys }}+\Delta S_{\text {surr }}\)
In the case of condensation of water, \(\Delta S_{\text {univ }}=\Delta S_{\text {water }}+\Delta S_{\text {surr }}\)
- The positive ΔSsurr outweighs the negative ΔSwater and the total entropy increases.
- For all spontaneous processes, a stage is reached when the system attains equilibrium. A glass of hot water cools till it attains room temperature. It does not cool below room temperature. At this point, it is said to have attained equilibrium or, to be more precise, thermal equilibrium.
A gas expands till it fills the whole of the available space uniformly. Then no further expansion occurs. Again this is a state of equilibrium. At the point of equilibrium,
ΔSuniv = 0 (at equilibrium).
Having understood that entropy is a measure of disorder or randomness of a system, it may be easy to imagine that at absolute zero, the entropy of a perfectly crystalline substance would be zero. At any temperature above this, the entropy is positive and it increases with an increase in temperature.
For any substance Ssolid < Sliquid < Sgas
For any system the heat changes are reflected in the entropy of that system. Hence, let us find an expression relating entropy and heat. Consider an exothermic reaction. The heat released in an exothermic reaction flows to the surroundings and the heat lost by the system is equal to the heat gained by the surroundings.
qsurr = -qsys
This equation is valid even for an endothermic reaction. In the case of an exothermic reaction, the flow of heat into the surroundings increases its entropy, i.e.,
ΔSsurr>0.
The actual magnitude of ΔSsurr however, depends on the amount of heat released as well as the temperature. In fact, the effect of q on entropy is related to temperature. The addition of heat to colder surroundings has a greater effect on the entropy as compared to the effect produced on addition of heat to hotter surroundings. The effect of q is greater if the temperature is lower. This can be mathematically expressed as
⇒ \(\Delta S_{\text {surt }}=\frac{q_{\text {surr }}}{T}=-\frac{q_{\text {sys }}}{T} .\)….(1)
We know that at equilibrium \(\Delta S_{\text {undv }}=0=\Delta S_{\text {sys }}+\Delta S_{\text {surr }} \text {. }\)….. (2)
Therefore, form equations (1) and (2) we get \(\Delta S_{s y s}=\frac{+q_{s y s}}{T}\)
If the process is reversible, the change in entropy of the system is given by \(\Delta S_{r v}=\frac{q_{r v v}}{T}.\)
- You may wonder that if no real processes take place reversibly, what is the use of the above equation. Although transfer of heat between system and surroundings is almost impossible to achieve in a reversible manner, this idealised path is important for the definition of ΔS.
- Entropy is a state function and the value of ΔS is the same for a change from say any state A to any other state B, irrespective of the path taken. Whether the change is carried out in a reversible or an irreversible manner, ΔS is the same.
- Thus whatever may be the actual path taken, ΔS is calculated using qrev only. qrev is the heat associated with the process had it traversed a reversible path.
The overall criterion for spontaneity: To get back to what we were discussing earlier, the overall criterion for the spontaneity of a process is the resultant of two tendencies—
- The tendency towards minimum energy and
- The tendency towards maximum entropy. These two tendencies act independently of each other and may work in the same or opposite directions.
The resultant of the two tendencies is expressed by another thermodynamic function called Gibbs free energy (G). The free energy of a system is a measure of its capacity to do useful work. The enthalpy (H), entropy (S), and free energy (G) of a system are related by the following expression.
G = H-TS (here T is the absolute temperature).
At constant temperature and pressure, the change in free energy (AG) during a process can be obtained by the following equation.
ΔG = ΔH -TΔS…… Equation 7
- The change in free energy takes into account both the change in enthalpy and the change in entropy, and is the factor which determines the spontaneity of a process. You know that a process is possible if it is accompanied by a decrease in energy (ΔH negative) and an increase in entropy (ΔS positive).
- Some processes for which ΔH is positive are possible because they are accompanied by an increase in entropy (ΔS positive). Some processes for which ΔS is negative are possible because they are accompanied by a decrease in energy (ΔH negative).
- Obviously, the conditions that are most favourable for the occurrence of a process are a decrease in energy and an increase in entropy, i.e., when ΔH is negative and ΔS is positive.
This makes ΔG negative. Basically, the criterion for a spontaneous reaction or feasibility of a reaction is that ΔG should be negative. A spontaneous reaction is always accompanied by a decrease in free energy, i.e.,
ΔG < 0 for a spontaneous process.
The tendency of the system to undergo a change in the direction of decreasing free energy makes it reach a state when ΔG = 0 at some point. This is the equilibrium state.
Thus, ΔG > 0 for a nonspontaneous process.
- However, the reverse of a nonspontaneous process (for which ΔG is positive) is spontaneous.
- In order to understand the meaning of free energy for a process being zero, let us consider the simple case of water. Water freezes spontaneously at T<0°C and icc melts spontaneously at T>0°C. At T = 0°C and 1 atm pressure, the ΔG for melting of ice and freezing of water is zero.
- At this temperature, theoretically, ice should not melt and water should not freeze. This means the two phases, liquid and solid, may coexist in equilibrium with each other.
- We may also have chemical reactions for which ΔG = 0. As a spontaneous chemical reaction proceeds (ΔG < 0), reactants turn into products and ΔG becomes less negative. At a point ΔG becomes zero—no more products are formed nor are any reactants consumed. This is a state of equilibrium or in fact chemical equilibrium.
- Reactions with negative ΔG are called exergonic and those with positive ΔG are called endergonic reactions.
When is ΔG negative? It is worth finding an answer to this question because it will help us determine which processes are feasible and under what conditions. The figure represents graphically the relation between ΔH, ΔS, and ΔG.
- Obviously, when ΔH is negative and ΔS is positive, ΔG will be negative (line ‘a’)
- When ΔH is negative and ΔS, too, is negative, ΔG will be negative if the magnitude of ΔH is greater than that of TΔS (line ‘b’), i.e., below temperature Tb.
- When both ΔH and ΔS are positive, AG will be negative if the magnitude of TΔS is greater than that of ΔH (line ‘c’, i.e., above temperature Tc).
- When ΔH is positive, while ΔS is negative, ΔG will be positive and the reaction will not be spontaneous.
Effect of temperature on spontaneity If you think about the last two conditions for ΔG to be negative, it will strike you that temperature must be an important factor in determining the spontaneity of a reaction. Say, both ΔH and ΔS are negative for a process. If tire magnitude of ΔH is greater than that of TΔS, ΔG will be negative and the process will be feasible.
- It could be possible that this is true up to a certain value of T, beyond which TΔS becomes greater in magnitude than ΔH. Similarly, if both ΔH and ΔS are positive for a particular process, there could be a temperature below which the magnitude of TΔS is less than that of ΔH, and the process is not feasible.
- For an endothermic process, ΔH is positive, i.e., the energy factor opposes the process. For such a process to occur, TΔS must be positive and its magnitude must be greater than that of ΔH.
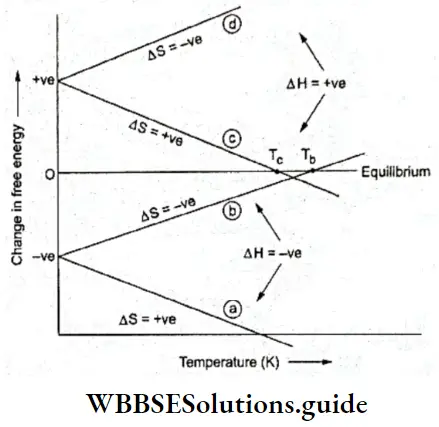
- For such a process, a higher temperature is more favorable as tills increases the magnitude of TΔS. In fact, there may be a temperature below which the magnitude of TΔS becomes less than that of ΔH and the process becomes unfeasible.
- For an exothermic process, ΔH is negative and ΔG is negative at all temperatures if ΔS is positive. However, if ΔS is negative, the process is feasible only if the magnitude of TΔS is less than that of ΔH.
- Obviously, such a reaction would be more feasible at a low temperature and may even become nonspontaneous above a particular temperature.
If ΔH is the major factor contributing to negative ΔG value, the reaction is called ‘enthalpy driven’ and if entropy is the factor behind it, it is called an ‘entropy driven’ reaction. A few values of \(\Delta H^{\ominus}, \Delta G^{\ominus} \text { and } \Delta S^\ominus\) are given in Table. Let us now summarise whatever we have discussed so far pertaining to the feasibility of a reaction in Table.
Units of \(\Delta G^{\ominus}\): cal mol-1 in the CGS system and J mol-1 or kJ mol-1 in the SI system.
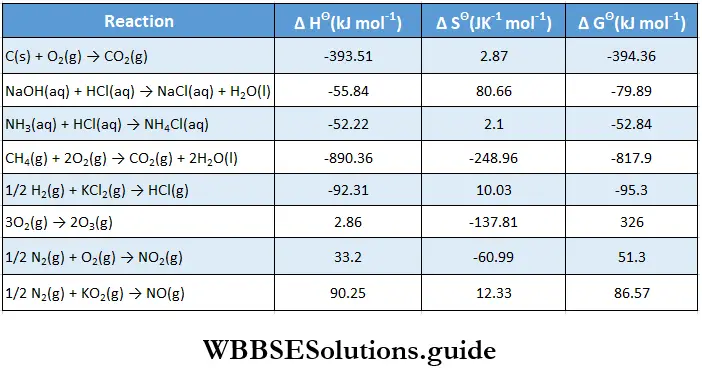
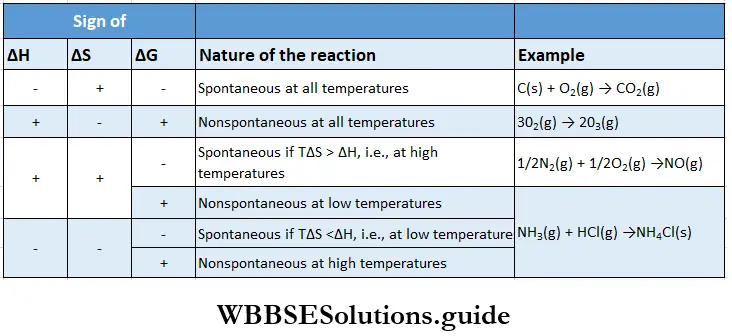
Standard free energy change: The standard free energy change of a reaction is defined as the free energy change at a specified temperature when the reactants in their standard states are converted to products in their standard states. It is denoted by \(\Delta G^{\ominus}\).
Calculation of \(\Delta G^{\ominus}\)° of a reaction Free energy is a state function. Hence, similar to \(\Delta H^{\ominus}\), \(\Delta G^{\ominus}\) for a reaction can be calculated from the free energy of formation of reactants and products.
The standard free energy of formation of an element in its standard state is taken as zero. Table lists values of standard enthalpy of formation, the standard free energy of formation, and absolute standard entropies of some compounds.
Tire standard free energy change of a reaction is obtained by subtracting the sum of the standard free energy of formation of the reactants from that of the products.
∴ \(\Delta_r G^{\ominus}=\Sigma v \Delta_f G^{\ominus} \text { (products) }-\Sigma v \Delta_f G^{\ominus} \text { (reactants) }\)
where v’s are the stoichiometric coefficients of the products and reactants respectively.
∴ \(\Delta G^{\ominus}\) may also be calculated from \(\Delta H^{\ominus}\) and \(\Delta S^{\ominus}\) of the reaction using an equation similar to Equation 7.
∴ \(\Delta_f G^{\ominus}\) = \(\Delta_f G^{\ominus}\) at a particular temperature.
We have seen how to calculate \(\Delta G^{\ominus}\) for a reaction. However, all chemical reactions do not occur with the reactants and products in their standard states. We can determine AG for a reaction of this kind using \(\Delta G^{\ominus}\) of the same reaction at standard conditions and the reaction quotient of the reaction. Consider the reaction
αA + βB → γC + δD
A, B, C, and D are the reactants and products while α,β,γ, and δ represent the respective stoichiometries.
The reaction quotient, Qc, of this reaction is given by \(Q_c=\frac{[C]^\gamma[D]^8}{[A]^\alpha[B]^\beta}\)
The square brackets indicate the concentrations of the reactant/product. Note that the products appear in the numerator and reactants appear in the denominator. The concentration terms of the species are raised to their respective stoichiometric coefficients.
ΔG at any temperature T is related to \(\Delta G^{\ominus}\) as follows:
ΔG = \(\Delta G^{\ominus}\) + RT In Qc ……. Equation 8
where R is the gas constant.
If the species involved in the reaction are gaseous in nature, concentration is replaced by partial pressure, and the reaction quotient is represented as Qp.
At equilibrium, Q = K, the equilibrium constant. (Qc = Kc or Qp = Kp, as the case may be.)
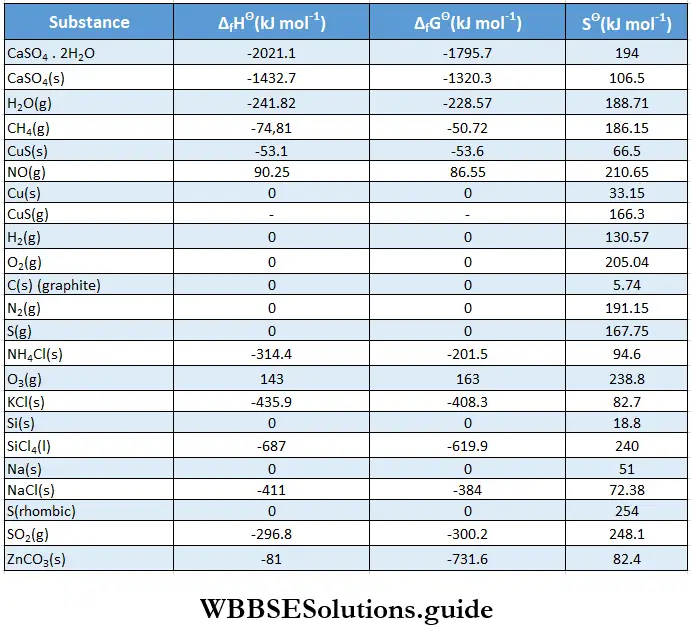
The Equilibrium constant K of the reaction αA + βB → γC + δD is given by
K = \(\frac{[\mathrm{C}]_e^r[\mathrm{D}]_e^5}{[\mathrm{~A}]_e^\alpha[\mathrm{B}]_e^\beta}\)
where the subscripts e denote equilibrium concentrations. Also, at equilibrium, ΔG = 0.
Hence, \(\Delta G^{\ominus}\) = -RT In K = -2.303 RT log K.
This gives yet another method of determining \(\Delta G^{\ominus}\) of a reaction. The pressure should be expressed in bar and the concentration in mol L-1 while determining Kp and Kc respectively.
Example 1. Calculate the entropy change AS per mole for the following reactions.
1. Combustion of hydrogen at 298 K \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
⇒ \(\Delta H=-241.6 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
⇒ \(\Delta G=-228.4 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
2. Vaporisation of methanol at its normal boiling point
Methanol (l) → Methanol (g), Δvap H = 23.9 kJ mol-1, Boiling point = 338 K
Solution:
1. ΔG = ΔH – TΔS.
∴ \(\Delta S=\frac{\Delta H-\Delta G}{T}=\frac{-2416-(-228.4)}{298}=-0.044 \mathrm{~kJ} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} .\)
2. ΔG = ΔH -TΔS.
ΔG = 0.
∴ \(\Delta S=\frac{\Delta_{\mathrm{vap}} H}{T}=\frac{23.9}{338}=0.071 \mathrm{~kJ} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} \text {. }\)
Example 2. Calculate the free energy change per mole for the following reactions and predict the feasibility of the reactions.
1. \(\mathrm{CaCO}_3(\mathrm{~s}) \longrightarrow \mathrm{CaO}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g}) \quad \text { at } 298 \mathrm{~K}\)
⇒ \(\Delta H=177.9 \mathrm{~kJ} \mathrm{~mol}^{-1} \quad \Delta S=160.4 \mathrm{JK}^{-1} \mathrm{~mol}^{-1}\)
2. \(2 \mathrm{NO}_2(\mathrm{~g}) \longrightarrow \mathrm{N}_2 \mathrm{O}_4(\mathrm{~g}) \quad \text { at } 298 \mathrm{~K}\)
⇒ \(\Delta \mathrm{S}=175.6 \mathrm{JK}^{-1} \mathrm{~mol}^{-1} \quad \Delta H=-57.2 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Solution:
1. ΔG =ΔH – TΔS.
∴ ΔG = 177.9 x 103 – 298 x 160.4 = 130100.8 J mol-1 = 130.1 kJ mol-1.
ΔG is positive. Therefore, the reaction is nonspontaneous at this temperature.
2. ΔG = -57.2×103 – (298 x 175.6) =-109528.8 J mol-1 =-109.5 kj mol”1.
ΔG is negative. Therefore, this reaction is spontaneous at 298 K.
Why Should There Be An Energy Crisis
While reading about the law of conservation of energy, it may have struck you as strange that there should be an energy crisis in the world when energy is not really lost in any process, it is merely converted from one form into another. It is true that energy is merely converted from one form into another, but it is useful to us only in certain forms.
- In most processes that we encounter, a portion of energy is converted into forms we cannot use. Let us look at it in another way. Most of our energy requirements are met by the burning of fossil fuels. The combustion of fuels produces energy which we use. But it also produces carbon dioxide and water, which escape into the air.
- The combustion of fuels is a spontaneous process, but the reverse is not a spontaneous process. So, the combustion of fuels is a one-sided process, which increases disorder. It is this tendency of most processes to increase disorder that wastes energy in our terms.
- Even the energy released during the combustion of fuels cannot be utilised fully to perform useful work. A part of it goes to increase disorder or is wasted according to us. This is why the efficiency of an engine is always less than 1.
∴ \(\frac{\text { work done by engine }}{\text { energy fed into engine }}<1\)
This follows from the equation
ΔG = ΔH-TΔS
or ΔH = ΔG + TΔS.
- If ΔH is the total energy fed into an engine then some of it, i.e., TΔS, will be used to increase disorder and only a part of it (ΔG) will be available for useful work. This is why, though the total energy of the world remains constant, every living process converts a part of it into a form that cannot be used.
- This is the part that goes to increase disorder in the universe. Since AG is the useful part of energy that can be harnessed, it is a measure of the maximum amount of energy that is ‘free’ to do useful work. Hence its name.
Example 1. Calculate the standard enthalpy of formation of ethyl alcohol from the following data.
- \(\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}(\mathrm{l})+3 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}_2(\mathrm{~g})+3 \mathrm{H}_2 \mathrm{O}(\mathrm{l}) \Delta_{\mathrm{f}} \mathrm{H}_1^{\ominus}=-1368 \mathrm{~kJ} \mathrm{~mol}^{-1}\) ….. (1)
- \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \Delta_{\mathrm{f}} H_2^{\ominus}=-393.5 \mathrm{~kJ} \mathrm{~mol}^{-1}\) …..(2)
- \(\mathrm{H}_2(\mathrm{~g})+\mathrm{S}_2 \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l}) \Delta_{\mathrm{f}} H_3^{\ominus}=-286.0 \mathrm{~kJ} \mathrm{~mol}^{-1}\)…..(3)
Solution: The required equation is
∴ \(2 \mathrm{C}(\mathrm{s})+3 \mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2 \rightarrow \mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}(\mathrm{l}) \quad \Delta_{\mathrm{f}} \mathrm{H}^{\ominus}=\)
To obtain this equation, first multiply Equation (2) by 2 and Equation (3) by 3 and add the products to get Equation (4). Then subtract Equation (1) from Equation (4),

∴ \(\Delta_{\mathrm{f}} H^{\ominus}=\left[2 \times \Delta_f H_2^{\ominus}+3 \times \Delta_{\mathrm{f}} H_3^{\ominus}\right]-\Delta_{\mathrm{f}} H_1^{\ominus}\)
= [(2 x – 393.5) + (3 x – 286.0)] – [-1368] = -1645 +1368 = -277 kJ mol-1.
Example 2. Calculate the heat of formation of methane, given that the heats of combustion of methane, graphite and hydrogen are – 890, – 394, – 286 kJ mol-1 respectively.
Solution:
The required equation is \(\mathrm{C}(\mathrm{s})+2 \mathrm{H}_2(\mathrm{~g}) \longrightarrow \mathrm{CH}_4(\mathrm{~g}) \quad \Delta_{\mathrm{f}} \mathrm{H}=\text { ? }\)
The equation for the combustion of methane is \(\mathrm{CH}_4(\mathrm{~g})+2 \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g})+2 \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \quad \Delta_{\mathrm{c}} \mathrm{H}_1=-890 \mathrm{~kJ} \mathrm{~mol}^{-1}\) ….(1)
The equation for the combustion of graphite is \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \quad \Delta_{\mathrm{c}} \mathrm{H}_2=-394 \mathrm{~kJ} \mathrm{~mol}^{-1}\) …..(2)
The equation for the combustion of hydrogen is \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2 \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \quad \Delta_{\mathrm{c}} \mathrm{H}_3=-286 \mathrm{~kJ} \mathrm{~mol}^{-1}\)…. (3)
Multiply Equation (3) by 2 and add the product to Equation (2).

This is the required equation.
Then ΔfH =[ΔcH2 + (2 X ΔcH3)]-[ΔcH1]
= ((-394) + (2 x – 286)] – [-890] = -394-572 + 890 = -76 kJ mol-1.
Example 3. Calculate the heat of reaction of \(\mathrm{CO}_2(\mathrm{~g})+\mathrm{H}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}(\mathrm{g})+\mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
Given that
- \(\mathrm{C}(\mathrm{s})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}(\mathrm{g}) \Delta_f H_2=-110.35 \mathrm{~kJ}\)
- \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{CO}_2(\mathrm{~g}) \Delta_f H_2=-393.35 \mathrm{~kJ}\)
- \(\mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l}) \Delta_f H_3=-241.6 \mathrm{~kJ}\)
Solution:
This problem can be solved by two methods.
1. Applying Hess’s law:
Add Equation (1) and Equation (3) and then subtract Equation (2) from the resultant equation.
⇒ \(\begin{aligned}
\mathrm{C}(\mathrm{s})+\mathrm{H}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) & \longrightarrow \mathrm{CO}(\mathrm{g})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \\
-\mathrm{C}\left(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g})\right. & \left.\longrightarrow \mathrm{CO}_2(\mathrm{~g})\right) \\
\hline \mathrm{H}_2(\mathrm{~g})+\mathrm{CO}_2(\mathrm{~g}) \longrightarrow & \longrightarrow \mathrm{CO}(\mathrm{g})+\mathrm{H}_2 \mathrm{O}(\mathrm{l})
\end{aligned}\)
This is the required equation.
∴ ΔrH = ΔfH1 – ΔfH3 – ΔfH2 =(-110.35) + (- 2416)- (-39335) = + 414 kj.
2. ΔfH = ∑ΔfH(products) – ∑ΔfH(reactants)
= [ΔfH(CO) + ΔfH(H2O)]- ΔfH(CO2)
= -110.35 + (- 2416)- (-39335) = + 414 kj.
Example 4. Just before an athletic event, a participant is given 100 g of glucose (C6H12O6), whose energy equivalent is 1560 kj. He utilises 50% of this gained energy in the event. Calculate the weight of water he would need to perspire in order to avoid storing extra energy in the body, ifthe enthalpy ofevaporation ofwater is 44 kj mol-1.
Solution:
Given
Just before an athletic event, a participant is given 100 g of glucose (C6H12O6), whose energy equivalent is 1560 kj. He utilises 50% of this gained energy in the event.
Energy from 100 g of glucose = 1560 kJ.
Energy utilised = 50% of1560 = 780 kJ.
Energy not used = 1560- 780 = 780 kJ.
Enthalpy of evaporation of water = 44 kJ mol-1.
44 kJ of energy evaporates1 mol of water.
780 kJ of energy would evaporate \(\frac{1 \times 780}{44}\) =17.73 mol of water.
17.73 mol of water = 17.73 x18 g = 319.14 g of water.
Example 5. Calculate the bond energy of the Cl—Cl bondfrom the equation \(\mathrm{CH}_4(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g}) \longrightarrow \mathrm{CH}_3 \mathrm{Cl}(\mathrm{g})+\mathrm{HCl}(\mathrm{g}) \Delta \mathrm{H}=-100.3 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
given that the bond energies ofC—H, C—Cl, H—Cl bonds are 413, 326 and 431 kJ mol-1 respectively.
Solution: The given equation can be written as
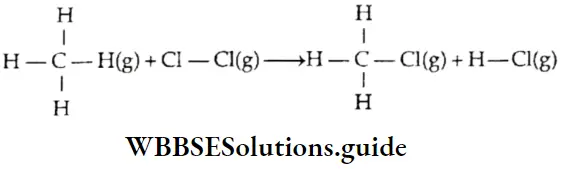
∴ \(\Delta H=\left[4 \Delta H_{\mathrm{C}-\mathrm{H}}+\Delta H_{\mathrm{C}-\mathrm{a}}\right]-\left[3 \Delta H_{\mathrm{C}-\mathrm{H}}+\Delta H_{\mathrm{C}-\mathrm{a}}+\Delta H_{\mathrm{H}-\mathrm{a}}\right]\)
or \(-100.3=\left[(4 \times 413)+\Delta H_{\mathrm{a}-\mathrm{a}}\right]-[(3 \times 413)+326+431]\)
∴ \(\quad \Delta H_{\mathrm{a}-\mathrm{C}}=243.7 \mathrm{~kJ} \mathrm{~mol}^{-1}\).
Example 6. Calculate the enthalpy change for the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{Br}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{HBr}(\mathrm{g})\) given that the bond enthalpies of H—H, Br—Br and H—Br are 435,192 and 364 kj mol-1 respectively.
Solution:
ΔH =∑ bond enthalpies of reactants – ∑ bond enthalpies of products
= [ΔHH-H + ΔHBr-Br] – [2 x ΔHH-Br]
= 435 + 192-(364 x 2)
= 627-728 =-101 kj mol-1.
Example 7. Calculate the Gibbs energy change for the reaction \(4 \mathrm{Fe}(\mathrm{s})+3 \mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{Fe}_2 \mathrm{O}_3(\mathrm{~s})\) at 25°C when O2 is at a partial pressure of 1.5 bar. \(\Delta_r G^e\) =-1484.4 kJ mol-1
Solution:
The reaction quotient Q = \(\frac{\left[\mathrm{Fe}_2 \mathrm{O}_3(\mathrm{~s})\right]^2}{[\mathrm{Fe}(\mathrm{s})]^4\left[p_{\mathrm{O}_2}\right]^3}\).
Since the concentrations of pure solids and liquids are constant,
Q = \(\frac{1}{\left(p_{\mathrm{O}_2}\right)^3}=\frac{1}{(1.5)^3}=0.2963\)
Now, \(\Delta_r G=\Delta_r G^\ominus\) + RT ln Q
= -1484.4 x 103 + 8.314 x 298 x ln (0.2963) (R = 8.314 JK-1 mol-1)
= – 1,484,400-3013.7 =-1,487,413.7 J mol-1 = -1487.4 kJ mol-1.
Example 8. Arrange the following systems in order of increasing entropy (compare one mole of each).
1. \(\mathrm{H}_2 \mathrm{O}$ (l), \mathrm{H}_2 \mathrm{O} (s), \mathrm{H}_2 \mathrm{O} (g)\)
2. \(\mathrm{H}_2(\mathrm{~g}), \mathrm{HBrO}_4(\mathrm{~g}), \mathrm{HBr}(\mathrm{g})\)
Solution:
Entropy varies as
1. \(\mathrm{H}_2 \mathrm{O}(\mathrm{s})<\mathrm{H}_2 \mathrm{O}(\mathrm{l})<\mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
2. \(\mathrm{H}_2(\mathrm{~g})<\mathrm{HBr}(\mathrm{g})<\mathrm{HBrO}_4(\mathrm{~g})\)
If the physical state is the same, the more complicated the molecule, the more is the entropy.
Example 9. For the reaction \(2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{SO}_3(\mathrm{~g})\) find ΔG at 25°C. The partial pressures of SO2, O2 and SO3 are 1.0 bar, 0.5 bar and 0.1 bar respectively. \(\Delta G^{\ominus}\) = -141.8kj mol-1.
Solution:
⇒ \(\Delta G=\Delta G^{\ominus}\) + RT In Qf; where Qf, is the reaction quotient.
⇒ \(Q_p=\frac{\left(p_{\mathrm{SO}_3}\right)^2}{\left(p_{\mathrm{SO}_2}\right)^2\left(p_{\mathrm{O}_2}\right)}=\frac{(0.1)^2}{(1)^2(0.5)}=0.02\)
∴ \(\Delta G=\Delta G^{\ominus}+R T \ln Q\)
= -1418 x 103 + 8314 x 298 x In 0.02
=-151,4923 J mol-1
=-151.5 kj mol-1
Example 10. The \(\Delta S_{\text {system }} \text { and } \Delta_t H^{\ominus}\) for the following reaction at 25 C are 220.5 J k-1 mol-1 and 90.7 kJ mol-1. Find S and say in which direction the reaction is not spontaneous.
⇒ \(\mathrm{CH}_3 \mathrm{OH}(\mathrm{g}) \rightleftharpoons \mathrm{CO}(\mathrm{g})+2 \mathrm{H}_2(\mathrm{~g})\)
Solution:
Given
The \(\Delta S_{\text {system }} \text { and } \Delta_t H^{\ominus}\) for the following reaction at 25 C are 220.5 J k-1 mol-1 and 90.7 kJ mol-1
⇒ \(\Delta S_{\text {total }}=\Delta S_{\text {sys }}+\Delta S_{\text {surr }} \text {. }\)
⇒ \(\Delta S_{\text {sys }}=220.8 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} \text {. }\)
⇒ \(\Delta S_{\text {surr }}=\frac{-\Delta H^{\ominus}}{T}\)
= \(-\frac{\left(90.7 \times 10^3\right)}{298} \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1}\)
= \(-304.4 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} \text {. }\)
\(\Delta S_{\text {total }}=220.8-304.4=-83.6 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1}\)As the entropy decreases (ΔS = -ve) the reaction is not spontaneous in the forward direction but the reverse reaction is spontaneous.
Example 11. Considering only entropy,find whether thefollowing reaction is spontaneous or not at 25°C
⇒ \(\mathrm{N}_2(\mathrm{~g})+2 \mathrm{H}_2(\mathrm{~g}) \rightleftharpoons \mathrm{N}_2 \mathrm{H}_4(\mathrm{l})\)
⇒ \(\mathrm{N}_2(\mathrm{~g}) \quad \mathrm{H}_2(\mathrm{~g}) \quad \mathrm{N}_2 \mathrm{H}_4(\mathrm{l})\)
Given \(\begin{array}{lccc}
\Delta H^{\ominus}\left(\mathrm{kJ} \mathrm{mol}^{-1}\right) & 0 & 0 & 50.6 \\
\Delta S^{\ominus}\left(\mathrm{J} \mathrm{K}^{-1} \mathrm{~mol}^{-1}\right) & 191.5 & 130.6 & 121.2
\end{array}\)
Solution: To predict whether or not a reaction is spontaneous, we must find the sign of \(\Delta S_{\text {total }}=\Delta S_{\text {sys }}+\Delta S_{\text {surr }}\)
The entropy change of the system is equal to the standard entropy of the reaction and
⇒ \(\Delta S_{\text {surr }}=-\Delta_r H^{\ominus} / T \text {. }\)
⇒ \(\Delta S_{\mathrm{sys}}=S^{\ominus}\left(\mathrm{N}_2 \mathrm{H}_4(\mathrm{l})\right)-S^{\ominus}\left(\mathrm{N}_2(\mathrm{~g})\right)-2 S^{\ominus}\left(\mathrm{H}_2(\mathrm{~g})\right)\)
= \(121.2-191.5-(2 \times 130.6)\)
= \(-3315 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} \text {. }\)
⇒ \(\Delta_{\mathrm{r}} H^{\ominus}=\Delta_f H^{\ominus}\left(\mathrm{N}_2 \mathrm{H}_4(\mathrm{l})\right)-\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{N}_2(\mathrm{~g})\right)-2 \Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{H}_2(\mathrm{~g})\right)\)
= \(50.6-0-0=50.6 \mathrm{~kJ}^{-1} \mathrm{~mol}^{-1} \text {. }\)
⇒ \(\Delta S_{\text {surr }}=-\frac{\Delta_r H^{\ominus}}{T}=\frac{-50.6}{25+273}=-0.1698 \mathrm{~kJ} \mathrm{~K}^{-1} \mathrm{~mol}^{-1}=-169.8 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} \text {. }\)
⇒ \(\Delta S_{\text {total }}=\Delta S_{\text {sys }}+\Delta S_{\text {surt }}\)
= \(-331.5+(-169.8)\)
“Thermodynamics, definition, laws, formulas, and solved examples”
= \(-5013 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1} \text {. }\)
As \(\Delta S_{\text {total }}\) is negative, the reaction is not spontaneous.
Example 12. The values of equilibrium constant for the following reaction at two different temperatures are 168 x 10-5 and 0.0123 respectively. If the first temperature is 1000°C, find the second temperature.
⇒ \(\mathrm{H}_2 \mathrm{O}(\mathrm{g})+\mathrm{C}(\mathrm{s}) \longrightarrow \mathrm{H}_2(\mathrm{~g})+\mathrm{CO}(\mathrm{g})\)
Solution:
Given
The values of equilibrium constant for the following reaction at two different temperatures are 168 x 10-5 and 0.0123 respectively. If the first temperature is 1000°C,
⇒ \(\Delta G^{\ominus}=-R T_1 \ln K_{p_1}\) …. (1)
⇒ \(\Delta G^{\ominus}=-R T_2 \ln K_{p_2}\)…. (2)
where T1 and T2 are the two different temperatures with the corresponding equilibrium constants \(K_{p_1}\) and \(K_{p_2}\) . Equating RHS of (1) and (2),
∴ \(R T_1 \ln K_{p_1}=R T_2 \ln K_{p_2}\)
or \(T_1 \ln K_{p_1}=T_2 \ln K_{p_2}\)
or \(T_2=T_1 \cdot \frac{\ln K_{p_1}}{\ln K_{p_2}}=(1273) \times \frac{\ln \left(168 \times 10^{-5}\right)}{\ln (0.0123)}=3182 \mathrm{~K}\) .
Example 13. Calculate Kp for thefollowing reactions at 298 K
- \(2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{SO}_2(\mathrm{~g}) \Delta G^{\ominus}=-142 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
- \(\mathrm{SO}_3(\mathrm{~g})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \longrightarrow \mathrm{H}_2 \mathrm{SO}_4(\mathrm{l}) \Delta G^{\ominus}=-81.7 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
Solution:
1. \(\Delta G^{\ominus}=-R^{\prime} T \ln K_r \) =\(-2.303 R T \log K_p\)
⇒ \(\log K_p=-\frac{\Delta G^{\ominus}}{2.303 R T}\)
⇒ \(\log K_p=-\frac{\Delta G^{\ominus}}{2.303 R T}\)
or \(K_p=\operatorname{Antilog}\left(\frac{-\Delta G^{\ominus}}{2.303 R T}\right)\)
= \(\text { Antilog }\left(\frac{142,000}{2.303 \times 8.314 \times 2.98}\right)=7.7 \times 10^{24}\).
2. \(K_p= Antilog \left(\frac{81,700}{2.303 \times 8.314 \times 298}\right)=2.08 \times 10^4\).
Example 14. Calculate \(\Delta G^{\ominus}\) for thefollowing reactions
1. \(\mathrm{CaCO},(\mathrm{s}) \longrightarrow \mathrm{CaO}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g})\)
⇒ \(K_p=1.06 \text { at } 500^{\circ} \mathrm{C}\)
2. \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\)
⇒ \(\mathrm{K}_p=98.9 \text { at } 300^{\circ} \mathrm{C}\).
3. \(\mathrm{CoO}(\mathrm{s})+\mathrm{CO}(\mathrm{g}) \rightleftharpoons \mathrm{Co}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g})\)
⇒ \(K_p=1.09 \times 10^4 \text { at } 277^{\circ} \mathrm{C}\) .
Solution: Since \(\Delta G^{\ominus}\), substituting the values of R, T and Kp we get \(\Delta G^{\ominus}\) for each reaction.
1. \(\Delta G^{\ominus}\) = -8.314 x (500 + 273) x ln(1.06) = -374.4 J mol-1.
2. \(\Delta G^{\ominus}\) = -8.314 x (300 + 273) x ln(98.9) = -21,885.9 J mol-1 = -21.88 kj mol-1.
3. \(\Delta G^{\ominus}\) = -8.314 x (277 + 273) x ln(1.09 x 104 )\(\Delta G^{\ominus}\)= -42310.2 J mol-1 = -42.51 kJ mol-1.
Example 15. Calculate \(\Delta G^{\ominus}\) for the following reactions which are carried out under standard state conditions.
- \(2 \mathrm{LiOH}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g}) \longrightarrow \mathrm{Li}_2 \mathrm{CO}_3(\mathrm{~s})+\mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
- \(\mathrm{H}_2(\mathrm{~g})+\mathrm{Br}_2(\mathrm{I}) \longrightarrow 2 \mathrm{HBr}(\mathrm{g})\)
- \(\mathrm{CaO}(\mathrm{s})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightarrow \mathrm{Ca}(\mathrm{OH})_2(\mathrm{~s})\)
- \(2 \mathrm{HgO}(\mathrm{s}, \text { red }) \rightarrow 2 \mathrm{Hg}(\mathrm{l})+\mathrm{O}_2(\mathrm{~g})\)
The standardfree energies of the compounds are given below.
⇒ \(\Delta_i G\left(\mathrm{~kJ} \mathrm{~mol}^{-1}\right)\)
⇒ \(\mathrm{LiOH}(\mathrm{s}) -443.9 \)
⇒ \(\mathrm{CO}_2(\mathrm{~g}) -394.4\)
⇒ \(\mathrm{Li}_2 \mathrm{CO}_3(\mathrm{~s}) -1132.4\)
⇒ \(\mathrm{H}_2 \mathrm{O}(\mathrm{g}) -228.6\)
⇒ \(\mathrm{H}_2(\mathrm{~g}) 0\)
⇒ \(\mathrm{Br}_2(\mathrm{l}) 0\)
⇒ \(\mathrm{HBr}(\mathrm{g}) -53.43\)
⇒ \(\mathrm{CaO}(\mathrm{s}) -604.2\)
⇒ \(\mathrm{H}_2 \mathrm{O}(\mathrm{l}) -237.2\)
⇒ \(\mathrm{Ca}(\mathrm{OH})_2(\mathrm{~s}) -896.8\)
⇒ \(\mathrm{HgO}(\mathrm{s}, \text { red) } -58.55\)
⇒ \(\mathrm{Hg}(\mathrm{l}) 0\)
⇒ \(\mathrm{O}_2(\mathrm{~g}) 0\)
⇒ \(\Delta_1 \mathrm{H}(\mathrm{Hg}(\mathrm{s}, \text { red }))=-90.83 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
⇒ \(\text { Substance } \mathrm{S}^{\ominus}\left(\mathrm{JK}^{-1} \mathrm{~mol}^{-1}\right)\)
⇒ \(\mathrm{Hg}_{\mathrm{g}} 38.01\)
⇒ \(\mathrm{O}_2(\mathrm{~g}) 205.04\)
⇒ \(\mathrm{HgO}(\mathrm{s}, \text { red) } 70.29\)
Which of these reactions islare spontaneous? For those reactions that are not spontaneous,find the temperature at which they will become spontaneous assuming ΔH and εS to be independent of temperature.
Solution:
1. \(\Delta G^{\ominus}=\Delta_f G^{\ominus}\left(\mathrm{Li}_2 \mathrm{CO}_3(\mathrm{~s})\right)+\Delta_{\mathrm{f}} G^{\ominus}\left(\mathrm{H}_2 \mathrm{O}(\mathrm{g})\right)-2 \times \Delta_f G^{\ominus}(\mathrm{LiOH}(\mathrm{s}))-\Delta_f G^{\ominus}\left(\mathrm{CO}_2(\mathrm{~g})\right)\)
= -1132.4 + (-228.6)- 2 x (-443.9)- (-3944)
= -78.8 kj Spontaneous reaction
2. \(\Delta G^{\ominus}=2 \times \Delta_f G^{\ominus}(\operatorname{HBr}(\mathrm{g}))-\Delta_f G^{\ominus}\left(\mathrm{H}_2(\mathrm{~g})\right)-\Delta_f G^{\ominus}\left(\mathrm{Br}_2(\mathrm{l})\right)\)
= 2 x -53.43- 0- 0
= -106.86 kj Spontaneous reaction
3. \(\Delta G^{\ominus}=\Delta_f G^{\ominus}\left(\mathrm{Ca}(\mathrm{OH})_2(\mathrm{~s})\right)-\Delta_{\mathrm{f}} G^{\ominus}(\mathrm{CaO}(\mathrm{s}))-\Delta_f G^{\ominus}\left(\mathrm{H}_2 \mathrm{O}(\mathrm{l})\right)\)
-896.8- (-6042)- (-237.2)
= -55.4 kjSpontaneous reaction
4. \(\Delta G^{\ominus}=2 \times \Delta_f G^{\ominus}(\mathrm{Hg}(\mathrm{l}))+\Delta_f G^{\ominus}\left(\mathrm{O}_2(\mathrm{~g})\right)-2 \times \Delta_f \mathrm{G}^{\ominus}(\mathrm{HgO}(\mathrm{s} ; \text { red }))\)
= 0 + 0 -(-585) = +58.5 kj.
As \(\Delta G^{\ominus}\) is positive, this reaction is not spontaneous. Now let us find \(\Delta H^{\ominus}\) and \(\Delta S^{\ominus}\).
⇒ \(\Delta H^{\ominus}=2 \times \Delta_{\mathrm{f}} H^{\ominus}(\mathrm{Hg}(\mathrm{l}))+\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{O}_2(\mathrm{~g})\right)-2 \times \Delta_{\mathrm{f}} H^{\ominus}(\mathrm{HgO}(\mathrm{s}, \mathrm{red}))\)
= 0 + 0- (-90.83) = 90.83 kj.
⇒ \(\Delta S^{\ominus}=2 \times S^{\ominus}(\mathrm{Hg}(\mathrm{l}))+S^{\ominus}\left(\mathrm{O}_2(\mathrm{~g})\right)-2 \times S^{\ominus}(\mathrm{HgO}(\mathrm{s}, \text { red }))\)
= (2 x 38.01) + 205.04- 2 x 70.29 = 140.47 JK-1
For the reaction to be spontaneous T\(\Delta S^{\ominus}\) should exceed \(\Delta H^{\ominus}\). Let us find the temperature at which T\(\Delta S^{\ominus}\) is equal to \(\Delta H^{\ominus}\). Assuming that \(\Delta H^{\ominus}\) and \(\Delta S^{\ominus}\) are independent of temperature.
⇒ T\(\Delta S^{\ominus}\) = \(\Delta H^{\ominus}\)
or T = \(\frac{\Delta H^{\ominus}}{\Delta S^{\ominus}}=\frac{90.83 \times 10^3}{140.47}\) = 646.6 K
Above 646.6 K, the reaction will be spontaneous because then T\(\Delta S^{\ominus}\) > \(\Delta H^{\ominus}\) and \(\Delta G^{\ominus}\) will be negative.
Example 16. Calculate \(\Delta S_295^{\ominus}\) for thefollowing phase changes.
- \(\mathrm{C}_6 \mathrm{H}_6(\mathrm{l}) \longrightarrow \mathrm{C}_6 \mathrm{H}_6(\mathrm{~g})\)
- \(\mathrm{Cu}(\mathrm{s}) \longrightarrow \mathrm{Cu}(\mathrm{g})\)
- \(\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
The standard entropies of C6H6(l), C6H6(g), Cu(s), Cu(g), H2O(l) and H2O(g) are 172.8, 269.2, 33.15, 1663, 69.91 and 188.71 JK-1 mol-1 respectively.
Solution:
1. \(\Delta S_{\text {vap }}^{\ominus} =S^{\ominus}\left(\mathrm{C}_6 \mathrm{H}_6(\mathrm{~g})\right)-S^{\ominus}\left(\mathrm{C}_6 \mathrm{H}_6(\mathrm{l})\right)\)
=269.2-172.8
∴ \(\Delta S_{\text {vap }}^{\ominus} =96.4 \mathrm{JK}^{-1}\)
2. \(\Delta S_{\text {sub }}^{\ominus} =S^{\ominus}(\mathrm{Cu}(\mathrm{g}))-S^{\ominus}(\mathrm{Cu}(\mathrm{s}))\)
=166.3-33.15
∴ \(\Delta S_{\text {sub }}^{\ominus} =133.15 \mathrm{JK}^{-1}\)
3. \(\Delta S_{\text {vap }}^{\ominus} \left.=S^{\ominus}\left(\mathrm{H}_2 \mathrm{O}(\mathrm{g})\right)-\mathrm{S}^{\ominus} \mathrm{H}_2 \mathrm{O}(\mathrm{l})\right)\)
=188.71-69.91
∴ \(\Delta S_{\text {vap }}^{\ominus} =118.8 \mathrm{JK}^{-1}\) .
Example 17. Calculate the standaril entropy changefor the combustion ofliquid ethanol, C2H5OH to gaseous CO2 and liquid water. The standard entropies of CO2(g), H2O(l), C2H5OH(l) and O2(g) are 213.6 K-1 mol-1, 161JK-1 mol-1 and 205.03 JK-1 mol-1 respectively.
Solution:
⇒ \(\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}(\mathrm{l})+3 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{CO}_2(\mathrm{~g})+3 \mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
⇒ \(\Delta S^{\ominus}=2 \times S^{\ominus}\left(\mathrm{CO}_2(\mathrm{~g})\right)+3 \times S^{\ominus}\left(\mathrm{H}_2 \mathrm{O}(\mathrm{l})\right)-S^{\ominus}\left(\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}\right)-3 \times\left(S^{\ominus}\left(\mathrm{O}_2(\mathrm{~g})\right)\right.\)
= 2 x 213.4 +3 x 69.91- 161-3 x 205.3 = -139.97 J K-1mol-1.
Example 18. Find the equilibrium constant of the reaction \(\mathrm{CH}_4(\mathrm{~g})+4 \mathrm{Cl}_2(\mathrm{~g}) \rightleftharpoons \mathrm{Cl}_4(\mathrm{~g})+4 \mathrm{HCl}(\mathrm{g})\)
The standard heats offormation of CH4 (g), CCl4 (g) and HCl(g) are -74.81 kJ mol-1,-102.9 kJ mol-1 and -92.31 kJ mol7 respectively. The standard molar entropies of CH4 (g), C2(g), CCl4(g), and HCl(g) are 186.26, 223.07, 309.7 and 186.91 J K-1 mol-1 respectively at 298 K.
Solution:
⇒ \(\Delta_{\mathrm{r}} H^{ominus}=\underset{\text { Products }}{\Sigma \Delta_{\mathrm{f}} H^{ominus}}-\underset{\text { Reactants }}{\Sigma \Delta_f H^{ominus}}\)
= \(\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{CCl}_4(\mathrm{~g})\right)+4 \Delta_{\mathrm{f}} H^{\ominus}(\mathrm{HCl}(\mathrm{g}))-\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{CH}_4(\mathrm{~g})\right)-4 \Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{Cl}_2(\mathrm{~g})\right)\)
= -102. 9 + (4 x -92.31)- (-74.81)
Since the standard enthalpies of formation of elements in their reference states are zero by definition
⇒ \(\Delta_{\mathrm{f}} H^{\ominus}\left(\mathrm{Cl}_2(\mathrm{~g})\right) \text { is } 0\)
⇒ \(\Delta_{\mathrm{r}} H^{\ominus} =-397.33 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
∴ \(\Delta_{\mathrm{r} S^{\ominus}} =\underset{\text { Reaction }}{\Sigma S^{\ominus}}-\underset{\text { Products }}{\Sigma S^{\ominus}}\)
= \(S^{\ominus}\left(\mathrm{CCl}_4(\mathrm{~g})\right)+4 S^{\ominus}(\mathrm{HCl}(\mathrm{g}))-S^{\ominus}\left(\mathrm{CH}_4(\mathrm{~g})\right)-4 S^{\ominus}\left(\mathrm{Cl}_2(\mathrm{~g})\right)\)
= 309.7 + (4 x186.91)- 186.26- (4 x 223.07) = -21.2 JK-1 mol-1
Substituting this value of \(\Delta_{\mathrm{r}} S \text { in } \Delta G^{\ominus}=\Delta H^{\ominus}-T \Delta S^{\ominus}\)
⇒ \(\Delta G^{\ominus}\) = -39733 x 103 -298(-21.2) = -391.01 kJ mol-1 = -391.01 kJ mol-1
⇒ \(\Delta G^{\ominus}\) = -RT In K = -2.303 RT log K.
log K = \(-\frac{\Delta G^{\ominus}}{2303 R T}\)
∴ K = \({antilog}\left(\frac{-\Delta G^{\ominus}}{2.303 R T}\right)\)
= \({antilog}\left(\frac{391.01 \times 10^3}{2.303 \times 8.314 \times 298}\right)\)
= antilog (68.53) = 3.37×1068.
Example 19. The standard enthalpy changefor the reaction \(\mathrm{NH}_3(\mathrm{aq})+\mathrm{HCl}(\mathrm{aq}) \longrightarrow \mathrm{NH}_4 \mathrm{Cl}(\mathrm{aq})\) at 298 K is -52.22 kJ. The standard Gibbs energy change is -52.81 kJ. Find the standard molar entropy of NH3(aq) if that of HCl(aq) and NH4Cl(aq) are 56.5 JK-1 mol-1 and 169.9 J K-1 mol-1 respectively.
Solution:
Given
The standard enthalpy changefor the reaction \(\mathrm{NH}_3(\mathrm{aq})+\mathrm{HCl}(\mathrm{aq}) \longrightarrow \mathrm{NH}_4 \mathrm{Cl}(\mathrm{aq})\) at 298 K is -52.22 kJ. The standard Gibbs energy change is -52.81 kJ.
⇒ \(\Delta G^{\ominus}\)=\(\Delta H^{\ominus}-T \Delta S^{\ominus}\)
∴ \(T \Delta S^\ominus=\Delta H^\ominus-\Delta G^\ominus\)
or, \(\Delta S^{\ominus}=\frac{\Delta H^{\ominus}-\Delta G^{\ominus}}{T}\)
= \(\frac{-52220 \mathrm{~J}-(-52810 \mathrm{~J})}{298}=\frac{590}{298}=1.98 \mathrm{~J} \mathrm{~K}^{-1} \text {. }\)
Now \(\Delta S^{\ominus}=S_{\mathrm{NH}_4 \mathrm{Cl}(\mathrm{aq})}^{\ominus}-\left(S_{\mathrm{NH}_3(\mathrm{aq})}^{\ominus}+S_{\mathrm{HCl}(\mathrm{aq})}^{\ominus}\right)\).
Substituting the values, we get
1.098= \(169.9-\left(S_{\mathrm{NH}_3(q)}^{\ominus}+56.5\right)\)
∴ \(S_{\mathrm{NH}_3}^{\ominus}=169.9-56.5-198\)
= \(111.42 \mathrm{JK}^{-1} \mathrm{~mol}^{-1}\).
Example 20. The \(\Delta_{\text {fus }} H^\ominus \text { and } \Delta_{\text {fus }} S^\ominus \text { of } \mathrm{CCl}_4 \text { are } 2.5 \mathrm{~kJ} \mathrm{~mol}^{-1} \text { and } 9.99 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1}\) respectively at 298 K. Find the temperature at which solid CCl4 and its liquid are in equilibrium at 1 atm.
Solution:
Given
The \(\Delta_{\text {fus }} H^\ominus \text { and } \Delta_{\text {fus }} S^\ominus \text { of } \mathrm{CCl}_4 \text { are } 2.5 \mathrm{~kJ} \mathrm{~mol}^{-1} \text { and } 9.99 \mathrm{~J} \mathrm{~K}^{-1} \mathrm{~mol}^{-1}\) respectively at 298 K.
⇒ \(\Delta G^{\ominus}=\Delta H^{\ominus}-T \Delta S^{\ominus}\).
But at equilibrium, \(\Delta G^{\ominus}\) = 0.
∴ \(\Delta H^{\ominus}\) = T\(\Delta S^{\ominus}\)
or T = \(\frac{\Delta H^{\ominus}}{\Delta S^{\ominus}}\)=\(\frac{2.5 \times 10^3}{9.99}=250.2 \mathrm{~K}\)
Example 21. Consider the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{HCl}(\mathrm{g}).\)
The standard enthalpy change at 298 K is -186.62 kj. The standard molar entropies of H2(g), Cl2(g) and HCl(g) are respectively 130.684, 223.07 and 186.91 J K-1 mol-1 respectively. Calculate the standard Gibbs energy change at 298 Kfor the reaction.
Solution:
⇒ \(\Delta G^{\ominus}\) = \(\Delta H^{\ominus}\) – T \(\Delta S^{\ominus}\)
Given \(\Delta H^{\ominus}\) = -186.62 k] and T = 298 K
∴ \(\Delta S^{\ominus}\) = \(\Delta S^{\ominus}\)(products) – \(\Delta S^{\ominus}\)(reactants)
= \(2 \times S^{\ominus}(\mathrm{HCl}(\mathrm{g}))-S^{\ominus}\left(\mathrm{H}_2(\mathrm{~g})\right)-S^{\ominus}\left(\mathrm{Cl}_2(\mathrm{~g})\right)\)
= 2 x 186.91- 130.684- 223.07 = 20.07 JK-1.
Substituting this value of \(\Delta S^{\ominus}\), we get
⇒ \(\Delta G^{\ominus}\) = -186.62 x 103 – 298 x 20.07
= -186,620-5980.86 = -192,600.86 J.
The standard free energy change for the reaction is -192.6 kj.
Example 22. Predict whether the entropy of the following reactions wll increase or decrease when the reaction occurs.
- \(\mathrm{CaSO}_4 \cdot 2 \mathrm{H}_2 \mathrm{O}(\mathrm{s}) \longrightarrow \mathrm{CaSO}_4(\mathrm{~s})+2 \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
- \(\mathrm{CH}_4(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{C}(\mathrm{s})+2 \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
- \(\mathrm{Cu}(\mathrm{s})+\mathrm{S}(\mathrm{g}) \longrightarrow \mathrm{CuS}(\mathrm{s})\)
- \(\mathrm{N}_2(\mathrm{~g})+2 \mathrm{O}_2(\mathrm{~g}) \longrightarrow 2 \mathrm{NO}(\mathrm{g})\) .
Solution:
Verify your predictions by calculating \(\Delta S^{\ominus}\) using \(S^{\ominus}\) values.
1. In the reactant side we have only a solid whereas in the product side, we also have a gas apart from a solid. Since gases have large entropy, the entropy of system increases.
∴ \(\Delta S^{\ominus}=S^{\ominus}\left(\mathrm{CaSO}_4(\mathrm{~s})+2 \times S^{\ominus}\left(\mathrm{H}_2 \mathrm{O}(\mathrm{g})\right)-S^{\ominus}\left(\mathrm{CaSO}_4 \cdot 2 \mathrm{H}_2 \mathrm{O}\right)\right.\)
= 106.5 + 2 x 188.71-194.0 = 289.92 JK-1
2. The number of gas molecules (two) is the same on both the reactant and the product side.
Depending on the actual entropies of the substances, \(\Delta S^{\ominus}\) may increase or decrease but only slightly.
∴ \(\Delta S^{\ominus}=S^{\ominus}(\mathrm{C}(\mathrm{s}))+2 \times S^{\ominus}\left(\mathrm{H}_2 \mathrm{O}(\mathrm{g})\right)-S^{\ominus}\left(\mathrm{CH}_4(\mathrm{~g})\right)-S^{\ominus}\left(\mathrm{O}_2(\mathrm{~g})\right)\)
= 5.74 + 2 x188.71- 186.15- 205.03 = -8.02JK-1
3. One of the reactants is in the gaseous state but the only product is a solid. Hence, the entropy will decrease.
∴ \(\Delta S^{\ominus}=S^{\ominus}(\mathrm{CuS}(\mathrm{s}))-S^{\ominus}(\mathrm{Cu}(\mathrm{s}))-S^{\ominus}(\mathrm{S}(\mathrm{g}))\)
= 66.5- 33.15-167.75 =1344 J K-1.
4. The total number of gas molecules decreases from three to two as the reaction proceeds from left to right. This results in a decrease in entropy.
∴ \(\Delta S^{\ominus}=2 \times S^{\ominus}(\mathrm{NO}(\mathrm{g}))-S^{\ominus}\left(\mathrm{N}_2(\mathrm{~g})\right)-2 \times S^{\ominus}\left(\mathrm{O}_2(\mathrm{~g})\right)\)
= 2 x 210.65-1915- 2 x 205.03 = -180.26 JK-1
Thermodynamics Multiple Choice Questions
Question 1. A well-stoppered thermos flask containing some ice cubes is an example of a
- Closed system
- Open system
- Isolated system
- Nonthermodynamic system
Answer: 3. Isolated system
Question 2. Which of the following is correct in the context of an adiabatic process?
- pΔV = 0
- q = +W
- ΔU=q
- q = 0
Answer: 4. q = 0
Question 3. The enthalpy change for the reaction \(\mathrm{C}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g}) \rightarrow \mathrm{CO}_2(\mathrm{~g})\) is
- Positive
- Negative
- Zero
- None of these
Answer: 2. Negative
Question 4. The enthalpy of formation of ammonia is – 46.0 kJ mol-1 What is the enthalpy change for the following reaction?
⇒ \(2 \mathrm{NH}_3(\mathrm{~g}) \longrightarrow \mathrm{N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g})\)
- + 46.0 kj mol-1
- – 23.0 kj mol-1
- – 92.0 kj mol-1
- + 92.0 kj mol-1
Answer: 4. + 92.0 kj mol-1
Question 5. The heat released when 0.6 mol of HNO3 solution is mixed with 0.2 mol of NaOH is
- 57.0 kj
- 11.4 kj
- 28.5kj
- 34.9 kj
Answer: 2. 11.4 kj
Question 6. When NaCl dissolves in water, the entropy
- Increases
- Decreases
- Remains the same
- None of these
Answer: 1. Increases
Question 7. The enthalpy change for the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{1/2}\mathrm{O}_2(\mathrm{~g}) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g})\) is called
- Enthalpy of formation of water
- Enthalpy of combustion of hydrogen
- Enthalpy of vaporisation of water
- None of these
Answer: 1. Enthalpy of formation of water and 2. Enthalpy of combustion of hydrogen
Question 8. The energy required to dissociate 4 g of gaseous hydrogen into free gaseous atoms is 208 kcal at 25°C. The bond energy of the H—H bond is
- 104 kcal mol-1
- 10.4 kcal mol-1
- 208 kcal mol-1
- 416 kcal mol-1
Answer: 1. 104 kcal mol-1
Question 9. In which of the following does entropy decrease?
- Crystallisation of sugar from a solution
- Rusting of iron
- Melting of ice
- Vaporisation of camphor
Answer: 1. Crystallisation of sugar from a solution
Question 10. For the reaction X→y Y, ΔH and ΔS are negative at a certain temperature. This reaction will be spontaneous at
- Low temperature
- High temperature
- The same temperature
- Very high temperature
Answer: 1. Low temperature
Question 11. A spontaneous reaction is impossible if
- Both ΔH and ΔS are positive
- ΔH and ΔS are negative
- ΔH is positive and ΔS is negative
- ΔH is negative and ΔS is positive
Answer: 3. ΔH is positive and ΔS is negative
Question 12. When a solid melts, there is
- An increase in entropy
- An increase in enthalpy
- A decrease in internal energy
- Decrease in enthalpy
Answer: 1. An increase in entropy and 2. An increase in enthalpy
Question 13. Which of the following conditions are favourable for a spontaneous process?
- ΔH=-ve TΔS = + ve
- ΔH = -ve TΔS = -ve TΔS<ΔH
- ΔH=+ve TΔS = + ve TΔS<ΔH
- ΔH = +ve TΔS = + ve TΔS>ΔH
Answer: 1. ΔH=-ve TΔS = + ve,
2. ΔH = -ve TΔS = -ve TΔS<ΔH and
4. ΔH = +ve 7ΔS = + ve TΔS>ΔH
Question 14. The enthalpies of elements in their standard states are taken as zero. Thus, the enthalpy of formation of a compound
- Will always be positive
- Will always be negative
- Will always be zero
- May be negative or positive
Answer: 4. May be negative or positive
Question 15. The enthalpy of combustion of a substance
- Is always positive
- Is always negative
- Is numerically equal to the enthalpy of formation
- Cannot be predicted
Answer: 2. Is always negative
“Thermodynamics, energy conservation laws, and mathematical equations
Question 16. When ammonium chloride is dissolved in water, the solution becomes cold. The change is
- Exothermic
- Endothermic
- Super cooling
- None of these
Answer: 2. Endothermic