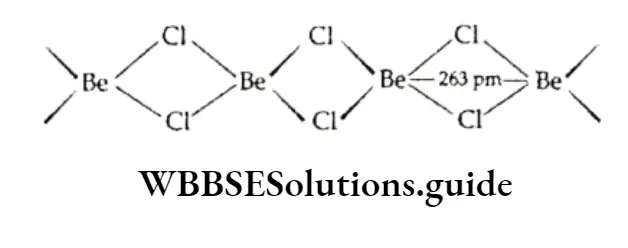
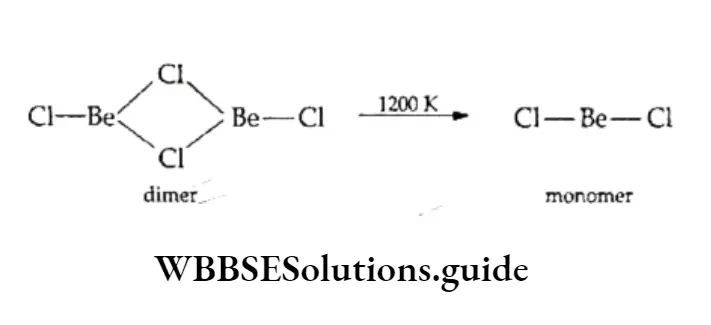
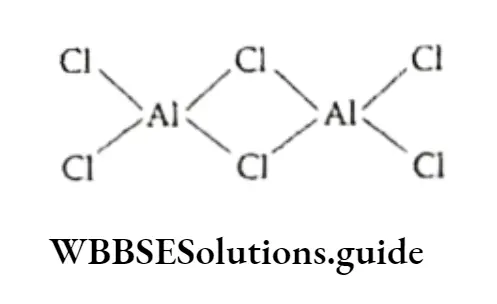
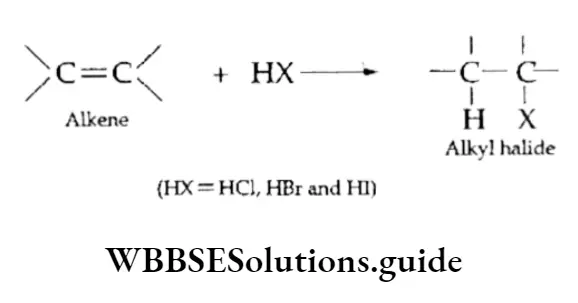
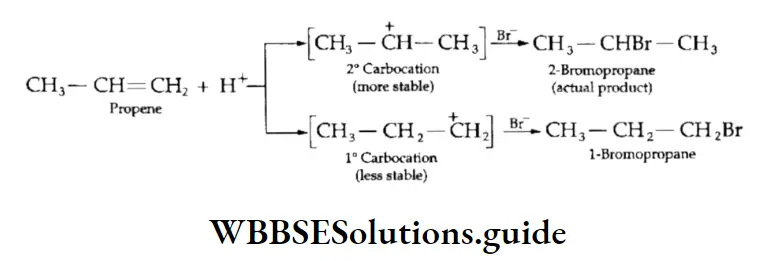
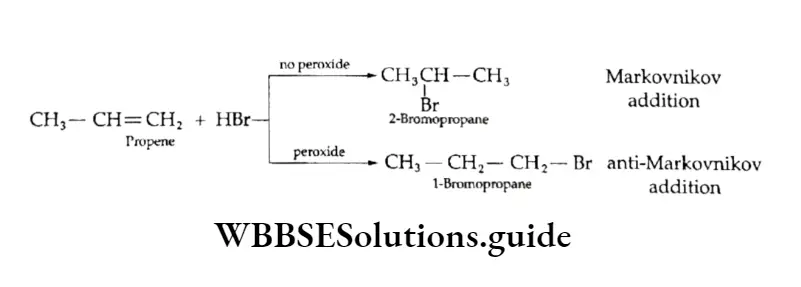
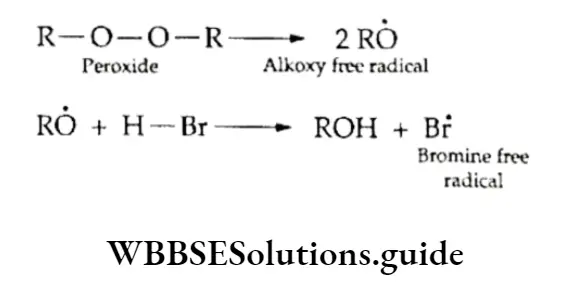
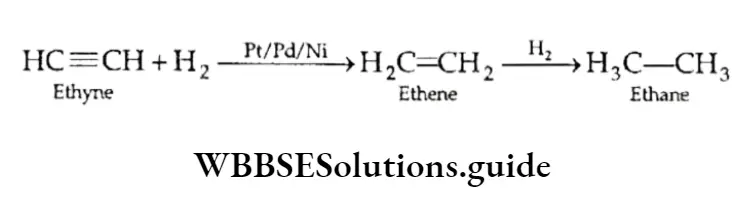
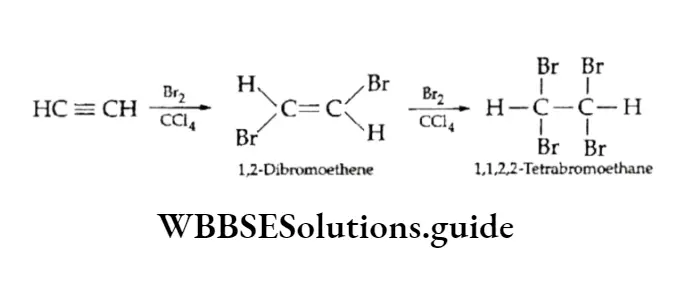
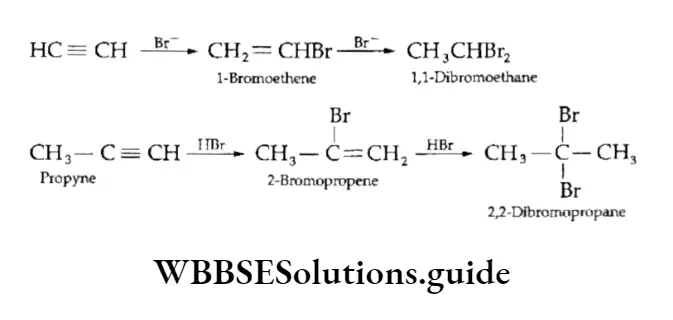
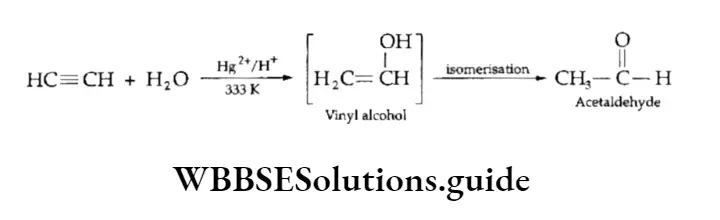
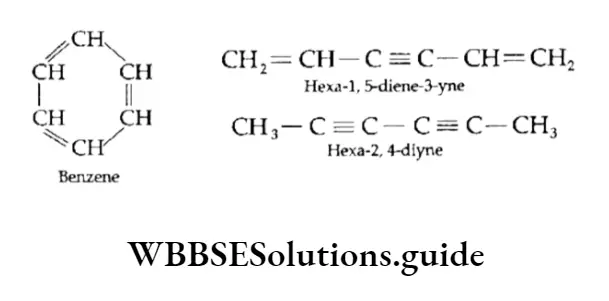
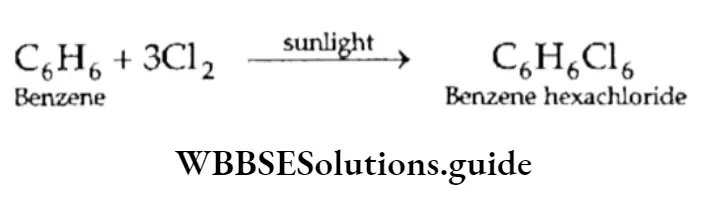
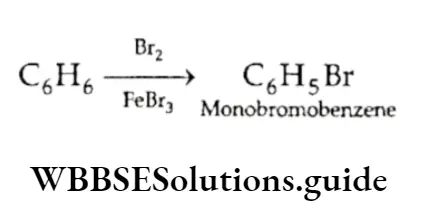
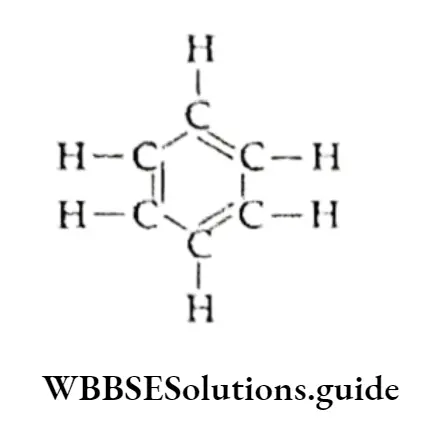
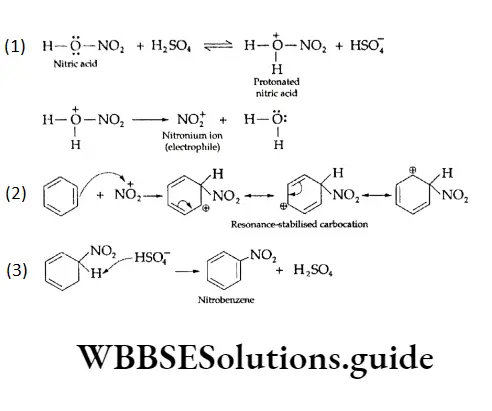
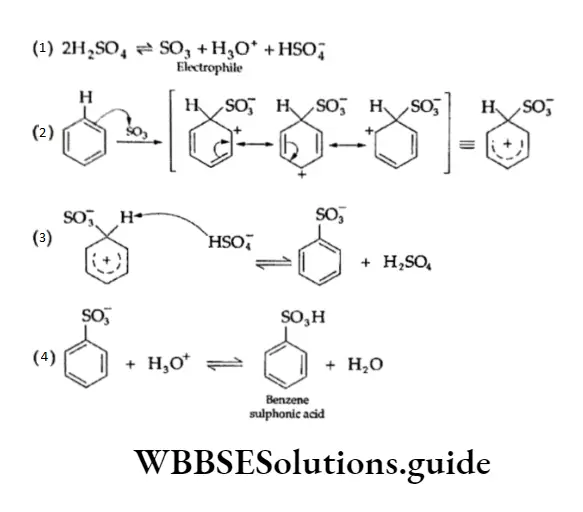
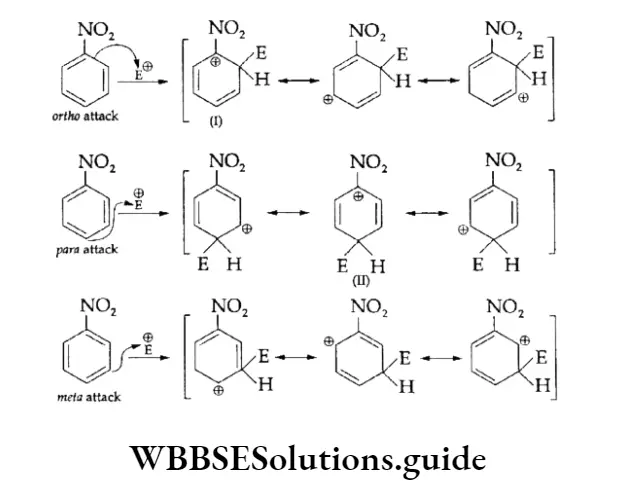
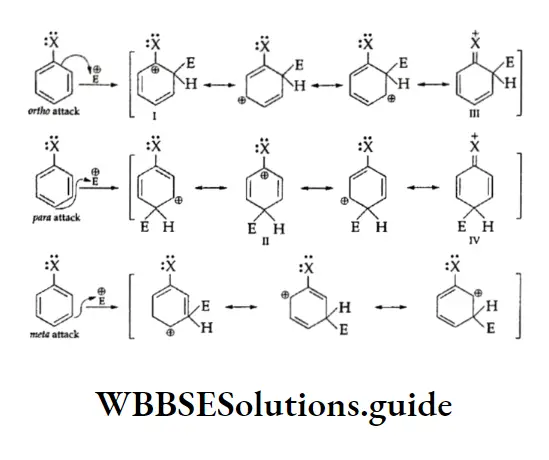
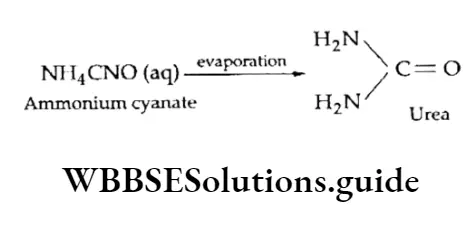
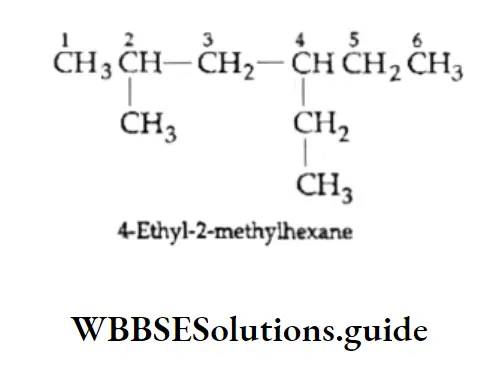
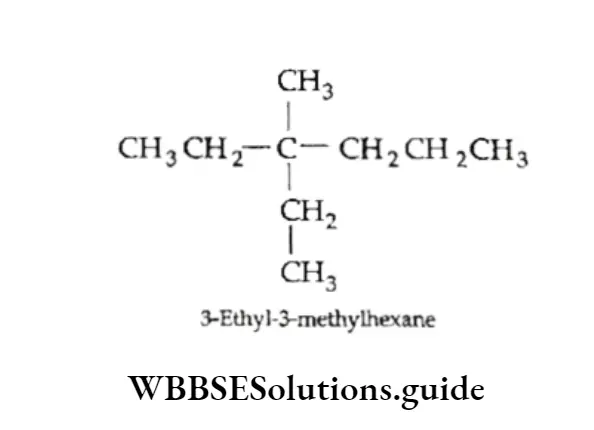
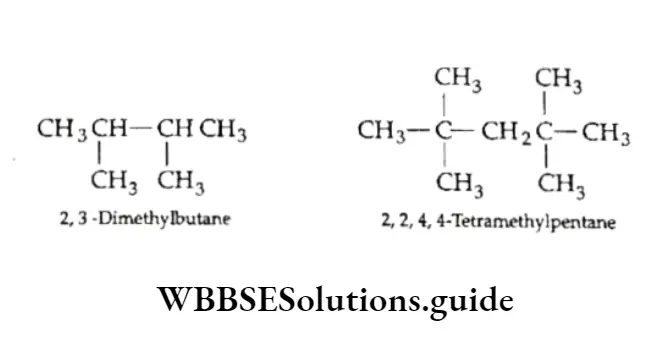
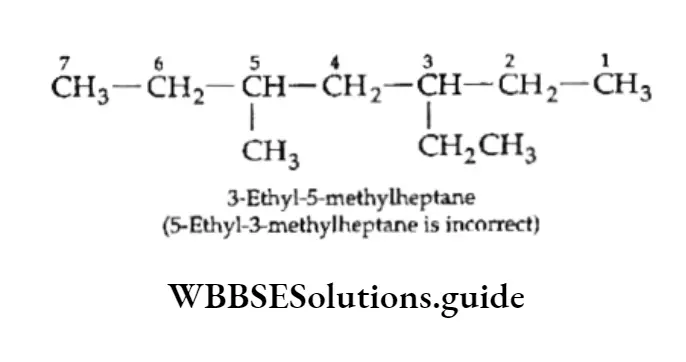
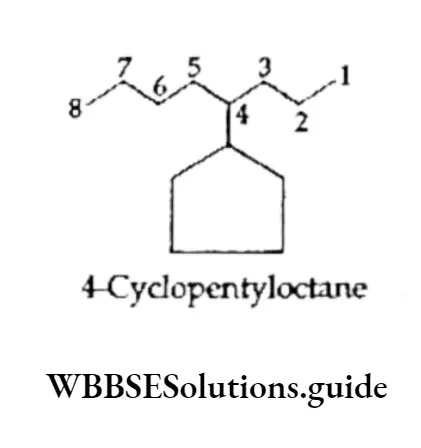
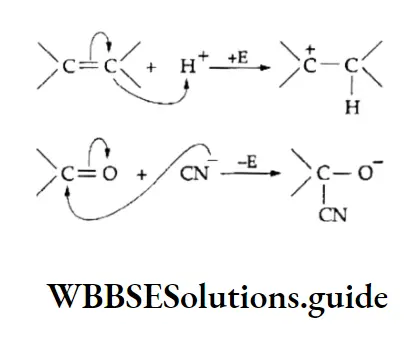
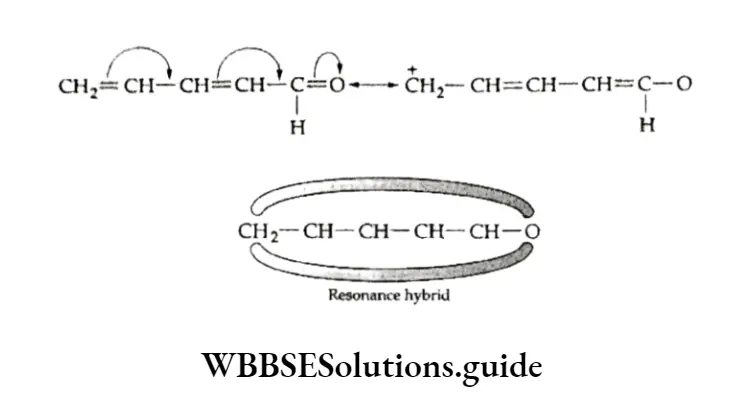
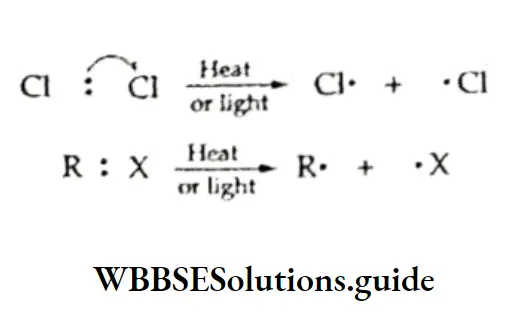
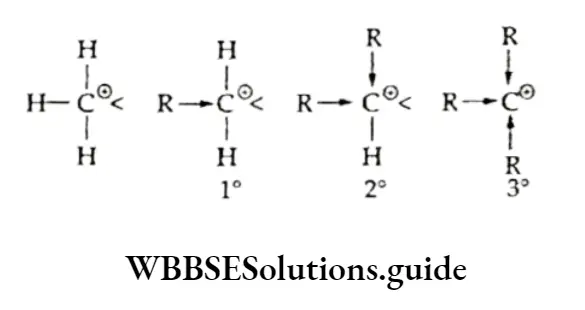
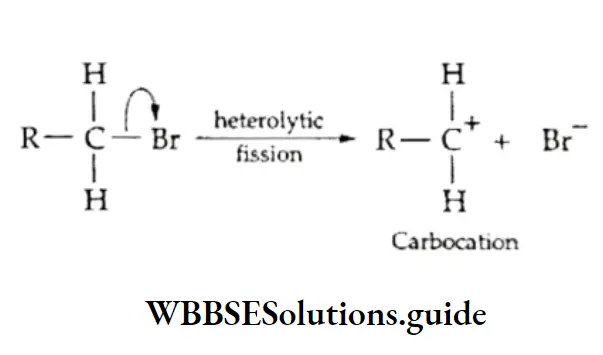
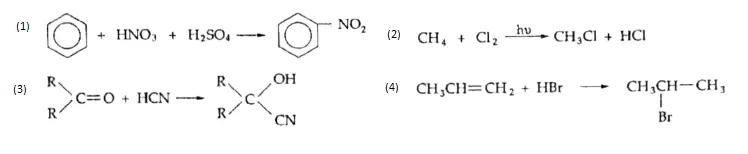
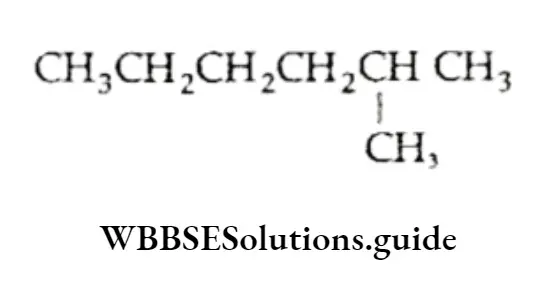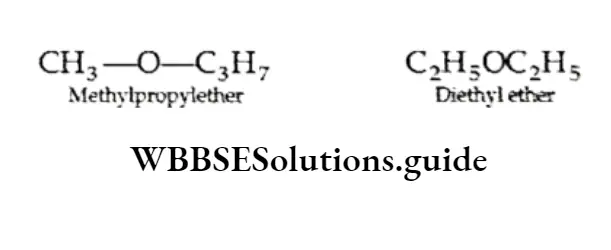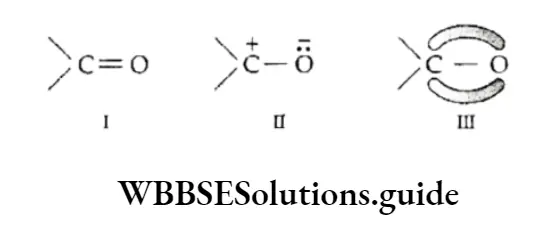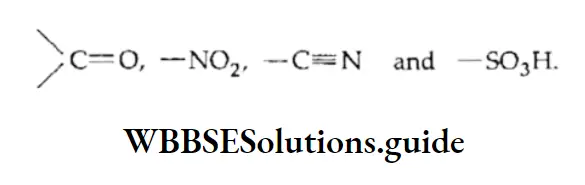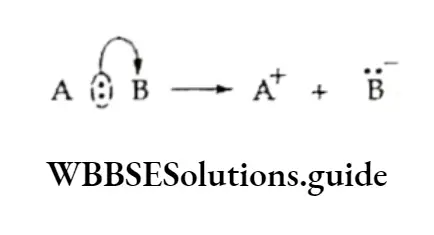Equilibrium
You have learnt about several aspects of a chemical reaction so far. You know, for example, that certain proportions of reactants react to form particular proportions of products and that these proportions are specified in a chemical equation. In the previous chapter, you learnt about the energy changes accompanying a chemical reaction. But there is one aspect of a chemical reaction that we have not considered at all.
Suppose we take reactants in exactly the required proportions (in the ratio of their respective number of moles in the balanced equation) and mix them together. Is it necessary that the reaction will proceed to completion? In other words, is it necessary that all of the reactants will be converted into products?
- We assume this in stoichiometric calculations (chemical arithmetic), but this is not always true. Quite often, a chemical reaction stops after a while and the resultant mixture contains reactants and products. It is said that the reaction has reached a state of chemical equilibrium.
- This state remains unchanged and the composition of the mixture remains the same, unless something forces a change. This something could be a change in temperature, pressure, or concentration of the reactants.
- It could even be a change in the concentration of the products, which could be achieved by removing the products (already formed) from the reaction site. These considerations, i.e., how to change the state of chemical equilibrium, are very important for industrial chemists. You can well imagine that the productivity of an industrial process may depend on these factors.
- When a reaction system reaches a chemical equilibrium, all the properties of the system, i.e., temperature, pressure, concentration of reactants and concentration of products, remain constant over time. What actually happens in such a system is that two opposing processes take place at the same time. Just as the reactants react to form the products, the products combine to produce the reactants.
And there comes a time when the forward process and the reverse process occur in such a way that the reaction seems to come to a standstill. At equilibrium, the rates at which the two opposing processes occur become equal.
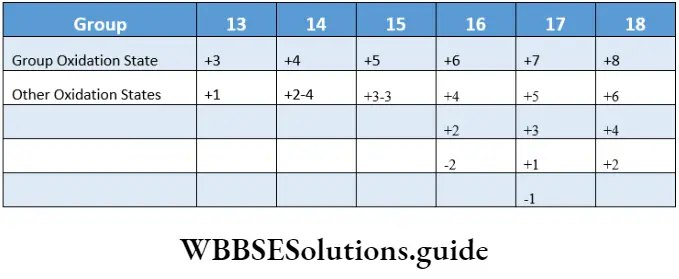
- A double arrow (\(\rightleftharpoons\)) is used to denote an equilibrium, whereas a single arrow (-») is used to denote a reaction which proceeds to completion. The extent to which the chemical reactions proceed may be different. Some reactions proceed nearly to completion and only a negligible concentration of the reactants is left.
- There are reactions in which the concentrations of reactants and products are comparable at equilibrium. Lastly, there are a few reactions in which most of the reactants remain unchanged at equilibrium, i.e., small amounts of products are formed.
- Our discussion so far has been centred around chemical reactions, so you may be tempted to believe that a state of equilibrium can be reached only in chemical processes. But this is not true.
- A state of equilibrium may be reached in a physical process as well. When the forward process and the reverse process occur simultaneously at exactly the same rate, a state of equilibrium is reached.

Equilibrium In Physical Processes
You have already come across physical processes which reach a state of equilibrium. A liquid in equilibrium with its own vapour inside a closed vessel is a system in which the processes of evaporation and condensation proceed at the same rate. The examples of physical equilibrium are solid \(\rightleftharpoons\)liquid, liquid \(\rightleftharpoons\) gas, solid \(\rightleftharpoons\) solution and gas solution.
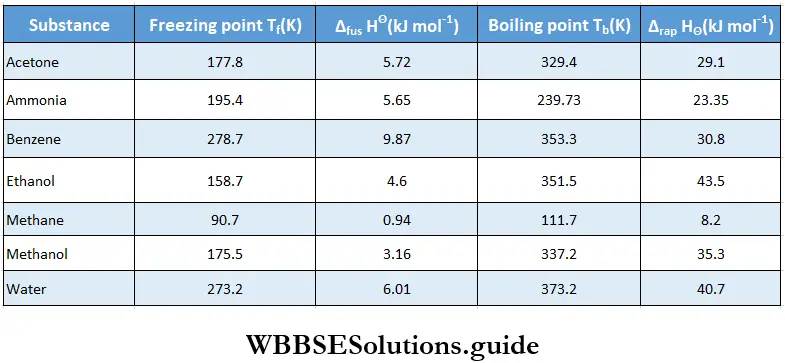
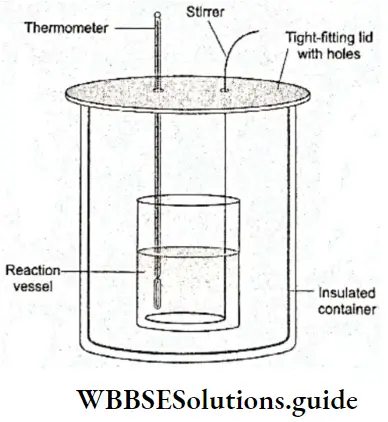
Solid-liquid equilibrium: If you drop a few ice cubes into a glass of water, all the ice changes into water after some time. This physical process or change proceeds in a particular direction until it is complete, i.e., until all the ice melts into water. Now suppose you pour a mixture of ice cubes and water at 273 K and normal atmospheric pressure into a perfectly insulated flask.
- The system, comprising the mixture of ice and water in the flask, will not exchange heat with the surroundings and a state of equilibrium will be reached, in which neither the temperature, the pressure, nor the composition of the mixture will change with time.
- It is not as though nothing will happen inside the flask, but there will be no way of telling that something is happening since there will be no outward indication.
- If you could become really small and enter the world of the molecules of water and ice, you would be able to see that what really happens is that molecules of ice go into the liquid state and molecules of water collide and stick to ice, constantly.
“Definition of equilibrium in chemistry with examples
The two processes go on simultaneously and at the same rate. Consequently, neither the mass of ice nor the mass of water changes.
To put this in terms of change in free energy, when an ice-water system is in equilibrium at 273 K and at 1 atm pressure, ΔG = 0. For the process, ice \(\rightleftharpoons\) water
ΔG < 0 when T > 273 K, and
ΔG > 0 when T < 273 K.
- The system comprising ice and water at 1 atm pressure can be at equilibrium only at 273 K. The solid and liquid phases of any (pure) substance at 1 atm pressure can be in a state of equilibrium only at a particular temperature.
- This temperature at which the solid and liquid phases of a pure substance are at equilibrium at a pressure of 1 atm is called the normal melting point or the normal freezing point of the substance.
The same definition but at a pressure of 1 bar refers to standard freezing point of a substance. The equilibrium between the solid and liquid phases of a substance at its melting point is a dynamic equilibrium, in which the activity in one direction is balanced by the activity in the reverse direction. Let us recall the characteristics of a system in dynamic equilibrium.
- The forward and reverse changes (processes) occur simultaneously and at the same rate, so that there is no change of mass of either side of the equilibrium.
- ΔG = 0
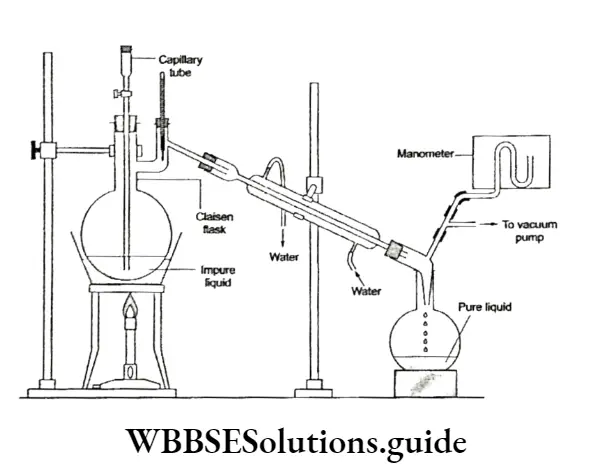
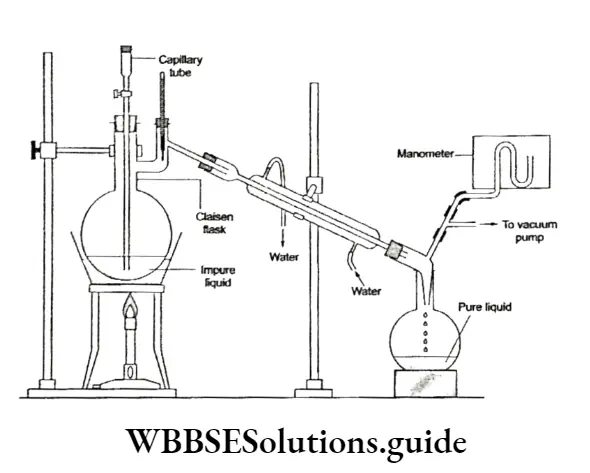
Liquid-vapour equilibrium: You have already come across this kind of equilibrium while studying vapour pressure. It can be demonstrated easily by performing a simple experiment. Take a small quantity of water at room temperature in a closed vessel connected to a manometer.
- Take care to see that the vessel has been evacuated first. To begin with, the level of mercury in both limbs of the manometer will be the ⇒ same. Then the level of mercury in the right limb of the manometer will rise slowly and the level of mercury in the left limb will fall.
- The level of water in the vessel falls gradually. There comes a time when the levels of mercury in tire two limbs become constant and so does the level of water in the vessel.
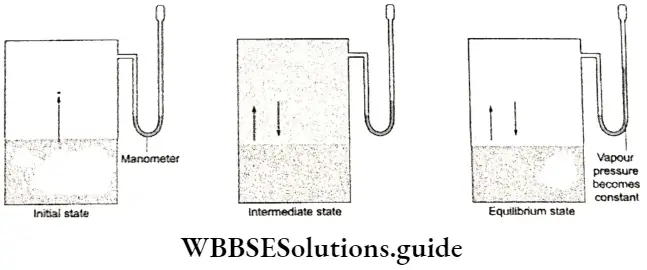
- Initially, there is no water vapour in the vessel and the level of mercury in both limbs is the same. As the water starts evaporating and more and more molecules escape into the gaseous phase, the pressure exerted by the water vapour increases, leading to a drop in the mercury level in the left limb and a rise in the right limb.
- But with the accumulation of vapour molecules in the vessel, the reverse process (condensation) starts. Some vapour molecules revert to the liquid phase. Condensation occurs at a lower rate than evaporation, to start with. But after a while, the two rates become equal and a state of equilibrium is established.
The pressure exerted by the vapour molecules becomes constant, as shown by the steady level of mercury in the manometer. The level of water in the vessel becomes constant because there is no net evaporation. The number of molecules entering the gas phase at any time is the same as the number of molecules entering the liquid phase.
“Types of equilibrium in chemistry with explanations”
∴ \(\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
The pressure exerted by the water vapour at equilibrium is called the equilibrium vapour pressure or simply the vapour pressure of water at the given temperature. In general, the pressure exerted by the vapour of a liquid in equilibrium with the liquid at a particular temperature is called the vapour pressure of the liquid at that temperature.
- Vapour pressure changes with temperature but is independent of the volume of the liquid. It does not depend on the volume of the container either.
- Different liquids have different vapour pressures at the same temperature and the liquid which has a higher vapour pressure is more volatile, or boils at a lower temperature. For instance, if the vapour pressure of liquid A is 3.2 kPa and that of another liquid B is 10.3 kPa at the same temperature, B is more volatile than A, or B has a lower boiling point than A.
- This should not be difficult to understand if you bear in mind that the vapour pressure of a liquid rises with temperature and that its boiling point is that temperature at which its vapour pressure becomes equal to the atmospheric pressure.
- Consider the two liquids A and B. The vapour pressure of B is higher than that of A at a particular temperature. As the temperature rises, the vapour pressures of both the liquids will rise.
Obviously, the vapour pressure of B will become equal to the atmospheric pressure at a temperature lower than that at which the vapour pressure of A will become equal to the atmospheric pressure. Consequently, the boiling point of B will be lower than that of A.
Solid-vapour equilibrium: In case of sublimable solids, the particles may escape occasionally into the vapour phase and establish vapour pressure.
- The particles which are at the surface of the solid and which have, at any point of time, higher average kinetic energy than the others, may escape into the vapour phase. If the solid is kept in a closed cylinder, particles are not able to escape and add to the vapour phase.
- Eventually, a point is reached when the particles from the vapour phase start returning to the solid phase. As in the case of liquid-vapour equilibrium initially, this reverse process is slow but after some time, an equilibrium is reached whereby rate of return of the particles to the solid phase becomes equal to the rate of escape.
- A dynamic equilibrium is thus established. The vapour pressure now corresponds to the equilibrium vapour pressure, which is characteristic of the solid.
- The tendency of the particles of the solid to escape depends on the intermolecular forces. Tire larger the intermolecular interaction, the less will be the vapour pressure of that solid.
Temperature also has a considerable effect on the vapour pressure of solids. The higher the temperature, the more energetic are the particles and the higher is the vapour pressure.
Iodine is a volatile solid. When solid iodine is placed in a closed vessel and the vessel is heated, the region above the solid becomes violet due to iodine vapours. The equilibrium is represented as \(\mathrm{I}_2 \text { (solid) } \rightleftharpoons \mathrm{I}_2 \text { (vapour). }\)
Another example is dry ice (solid CO2); it sublimes directly from solid to gas at atmospheric pressure.
- So far we have been considering closed systems. Systems we encounter in our everyday lives are (generally) not closed. The atmosphere, into which water vapour from lakes and rivers and other water bodies escapes, is an open system.
- Though the rate of evaporation of a liquid at a particular temperature in an open system is the same as that in a closed system, equilibrium can never be reached in an open system.
- This is because the rate of condensation can never be equal to the rate of evaporation in an open system, in which the vapour molecules get dispersed in a large volume.
- The water molecules which escape into the atmosphere, for instance, do not remain confined to a small space, they, get dispersed by the wind.
- How much water vapour is there in the air (what we refer to as humidity) depends on several factors, such as the presence of water bodies in the area, the temperature and the velocity of the wind.
- At places close to the sea, especially when the wind velocity is not high, the accumulation of water vapour in the air leads to acute discomfort.
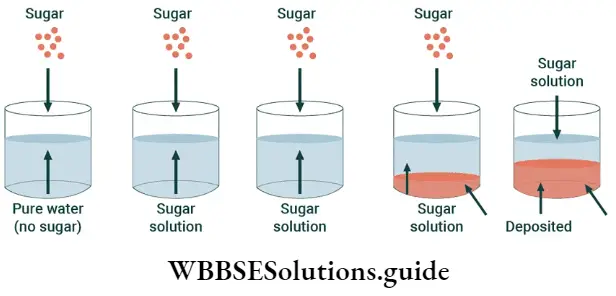
Solid-solution equilibrium: It is true that sugar dissolves in water. But you cannot dissolve an indefinite amount of sugar in a particular amount of water. You can, of course, dissolve more sugar in a particular amount of water if you heat the solution.
“Difference between static and dynamic equilibrium with examples”
- This is what they do in sweet shops when they prepare sugar syrups. But when the syrup cools, crystals of sugar separate from the solution because when the solution (syrup) cools, the solubility of the solute (sugar) in the solvent (water) decreases.
- You may remember from your science courses in junior classes that a solution in which more solute cannot be dissolved is called a saturated solution.
- The amount of a solute that has to be dissolved in a certain amount of a solvent to prepare a saturated solution at a particular temperature is called the solubility of the solute in that solvent at that temperature.
- Solubility is usually expressed in terms of the number of grams of a solute that dissolves in 100 mL of the solvent.
- The solubility of a solid in a liquid depends on temperature, but we will come to that later. Pressure has hardly any effect on the solubility of a solid in a liquid.
What happens when a solution becomes saturated is that a dynamic equilibrium is established between the molecules of the solute in the solid state and the molecules of the solute in the solution.
Solute (in solution) \(\rightleftharpoons solute\) (solid)
- The process by which molecules of the solid solute go into solution is called dissolution. The reverse process by which molecules of the solute in the solution revert to the solid state is called precipitation.
- When a solute is added to a solvent, there are no molecules of the solute in the solvent to begin with. Slowly, as the solute dissolves in the solvent, the reverse process begins.
At first, the process of dissolution proceeds at a faster rate than the process of precipitation. Than there comes a point when the two processes proceed at the same rate. This happens when equilibrium is reached.
Rate of dissolution = rate of precipitation
It is not as though activity comes to a standstill in the solution, but the number of molecules of the solute going into the solution (in a certain time) becomes the same as the number of molecules returning to the solid state.
- This can be demonstrated with the help of an experiment. Prepare a saturated sugar solution in a beaker in such a way that some undissolved sugar remains at the bottom of the beaker.
- If you add some sugar-containing radioactive carbon to the solution, in a while the solution and the undissolved sugar contains sugar which has radioactive carbon.
- This shows that though the solution is saturated, the process of dissolution continues and that the rate of dissolution must be equal to the rate of precipitation, since the amount of sugar left undissolved remains the same.
- In other words, the experiment demonstrates the dynamic nature of the equilibrium reached.
Gas-solution equilibrium: You must have heard people referring to drinks like Pepsi or Mirinda as carbonated or aerated water.
- Well, strictly speaking, such drinks also contain sugar and other additives and it would be more correct to refer to soda as aerated water.
- Nonetheless, it is true that drinks like Pepsi, too, have carbon dioxide dissolved under pressure. It is this carbon dioxide which escapes with a hissing sound when a bottle of coke or soda is opened.
- The fizz that you enjoy in such drinks is also because of the dissolved carbon dioxide.
- Aerated drinks are manufactured by dissolving carbon dioxide in them at a high pressure. Unlike solids, the solubility of gases (in liquids) increases with pressure.
The effect of pressure on the solubility of a gas in a liquid is given by Henry’s law (William Henry was a British physician and chemist), which says that the mass of a gas dissolved in a given mass of a solvent at a particular temperature is proportional to the pressure of the gas above the solvent.
- The solubility of a gas in a liquid decreases as temperature increases. When an aerated drink is bottled there is a dynamic equilibrium between the gas molecules above the solution (tire drink) and the gas molecules in the solution.
- This equilibrium holds as long as the pressure is maintained. When the bottle is opened, the pressure above the solution falls and some of the dissolved gas escapes to reach a new equilibrium.
- If an aerated drink is left open to the air for some time, it turns ‘flat’ because most of the dissolved gas escapes as the pressure above the drink (solution) becomes equal to the atmospheric pressure.
Henry’s law can be expressed mathematically as follows.
“Chemical equilibrium definition and types with MCQs”
∴ m∝ p or m = kp,
where m is the mass of the gas dissolved in the solution, p is the pressure of the gas above the solution and k is a constant of proportionality known as the Henry constant.
- Let us look at this another way. The mass of the gas dissolved in the solution will be related to the concentration of the gas in the solution.
- The pressure exerted by the gas above the solution will depend on the concentration of the gas above the solution. This gives us another way of expressing Henry’s law.
- Concentration of gas in solution x concentration of gas above solution
or \(\frac{\text { concentration of gas in solution }}{\text { concentration of gas in gaseous phase }}=\text { constant. }\)
Example: Suppose 0.30 g of iodine is stirred in 100 mL of water at 288 K until equilibrium is reached. Hoiv much iodine would dissolve in the water, if the solubility of iodine in water at 288 K is 0.0011 mol L-1 [Another way of expressing this is I2 (aq) at equilibrium = 0.0011 mol L-1 at 288 K]. Suppose another 100 mL of water is added to the solution after equilibrium is reached with 0.30 g of iodine and 100 mL of water. How much iodine would be left undissolved and what would be the concentration of iodine in the solution?
Solution:
Given
Suppose 0.30 g of iodine is stirred in 100 mL of water at 288 K until equilibrium is reached. Hoiv much iodine would dissolve in the water, if the solubility of iodine in water at 288 K is 0.0011 mol L-1 [Another way of expressing this is I2 (aq) at equilibrium = 0.0011 mol L-1 at 288 K]. Suppose another 100 mL of water is added to the solution after equilibrium is reached with 0.30 g of iodine and 100 mL of water.
The solubility of iodine (concentration at equilibrium) is given as 0.0011 mol L-1 at 288 K.
This means at equilibrium mass of iodine dissolved in 1 L of water = 0.0011 x 254 ≅ 0.279 g s 0.28 g [molar mass of I2 = 254 g]
∴ mass of iodine dissolved in 100 mL of water = 0.028 g.
If another 100 mL of water is added after equilibrium has been established, another 0.028 g of iodine would dissolve.
∴ mass of undissolved iodine – 0.30 – (0.028 x 2) = 0.30 – 0.056 = 0.244 g.
The concentration of iodine in the solution would simply be the solubility of iodine at equilibrium i. i.e., 0.28 gL-1.
Characteristics of equilibrium in physical processes: We have discussed the attainment of an equilibrium state in relation to four different physical processes. They all have certain characteristics in common.
- An equilibrium can be reached only in a dosed system, i.e., a system which neither gains matter from nor loses matter to the surroundings. For instance, in the case of a liquid—gas system, equilibrium is not reached if the vapour is allowed to escape from the vessel.
- The equilibrium reached is dynamic in nature, i.e., there is a balance between two opposing processes occurring at the same rate.
- At equilibrium, the measurable properties of the system become constant (under a given set of conditions). For instance, in the case of evaporation, the vapour pressure of the liquid is constant at a given temperature.
- In solid-liquid equilibrium, the melting point (the temperature at which the two phases coexist provided no heat is exchanged) is constant for a particular pressure.
- For the dissolution of a solid in a liquid, the solubility is constant at a given temperature. For the dissolution of a gas in a liquid, the concentration of the gas in the solution is constant for a particular pressure of the gas above the solution at a given temperature.
- At equilibrium, the concentrations of substances in the liquid and gaseous phase bear a constant ratio at a particular temperature. For the dissolution of C02 in water, for example, \(\frac{\mathrm{CO}_2(\mathrm{aq})}{\mathrm{CO}_2(\mathrm{~g})}\) is constant at a given temperature. This constant is called the equilibrium constant and its magnitude indicates the extent to which the process proceeds before equilibrium is reached. For example, the greater the value of \(\frac{\mathrm{CO}_2(\mathrm{aq})}{\mathrm{CO}_2(\mathrm{~g})}\) greater is the extent to which CO2 disiolves in water.
Equilibrium In Chemical Systems
We started this chapter with a discussion on whether a chemical reaction must always proceed to completion. There are some chemical reactions which proceed to completion, i.e., at equilibrium the concentration of the, reactants is negligible compared to the concentration of the products. (In reality, no reaction proceeds to completion in the true sense, but we need not concern ourselves with this now.) An example of this type of reaction is
⇒ \(\mathrm{BaCl}_2(\mathrm{aq})+\mathrm{Na}_2 \mathrm{SO}_4(\mathrm{aq}) \longrightarrow \mathrm{BaSO}_4(\mathrm{~s})+2 \mathrm{NaCl}(\mathrm{aq})\)
We could also call this reaction an irreversible reaction, since the reaction proceeds in one direction and the reverse process, i.e., the products reacting to form the reactants under the same conditions, does not take place. A few irreversible reactions are given below.
⇒ \(2 \mathrm{Na}+2 \mathrm{H}_2 \mathrm{O} \longrightarrow 2 \mathrm{NaOH}+\mathrm{H}_2\)
⇒ \(\mathrm{AgNO}_3(\mathrm{aq})+\mathrm{NaCl}(\mathrm{aq}) \longrightarrow \mathrm{NaNO}_3(\mathrm{aq})+\mathrm{AgCl}(\mathrm{s})\)
⇒ \(2 \mathrm{Mg}(\mathrm{s})+\mathrm{O}_2(\mathrm{~s}) \longrightarrow 2 \mathrm{MgO}(\mathrm{s})\)
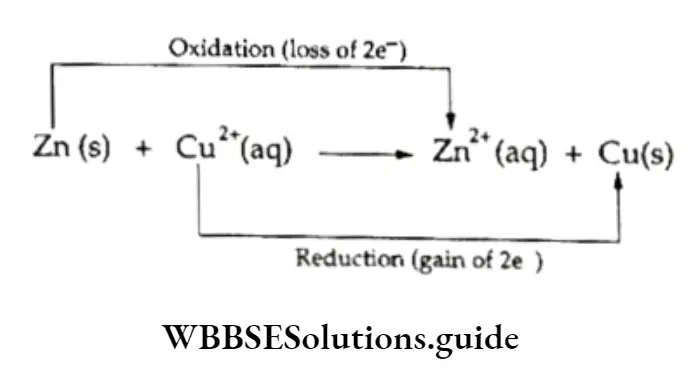
Let us now consider a reaction which is reversible, i.e., a reaction in which the reactants react to form products under certain conditions and the products combine to form the reactants under the same conditions.
For instance, iron reacts with steam to form hydrogen and iron oxide, and heated iron oxide reacts with hydrogen to produce iron and steam.
⇒ \(3 \mathrm{Fe}+4 \mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{Fe}_3 \mathrm{O}_4+4 \mathrm{H}_2\)
If a reversible reaction is carried out in such a way that one of the products escapes, the reaction becomes irreversible, or proceeds in only one direction.
- If, for instance, in the reaction between metallic iron and steam, the hydrogen formed is allowed to escape, the reverse reaction cannot take place and the process becomes virtually irreversible, or proceeds to completion.
- If, on the other hand, the reaction is carried out in a closed vessel, from which hydrogen cannot escape, both forward and backward reactions take place simultaneously and there comes a time when an equilibrium is established.
- At equilibrium, the reaction appears to have stopped, but what happens is that the forward and reverse reactions occur at the same rate.
For instance, when hydrogen and iodine are heated in a closed vessel at 717 K, they react to form hydrogen iodide. However, since the hydrogen iodide is not allowed to escape, the reverse reaction occurs. Hydrogen iodide decomposes to form hydrogen and iodine.
⇒ \(\underset{\text { colourless }}{\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g})} \rightleftharpoons \underset{\text { viclet }}{2 \mathrm{HI}(\mathrm{g})}\)
Initially, the reaction mixture is deep violet because of the presence of iodine.
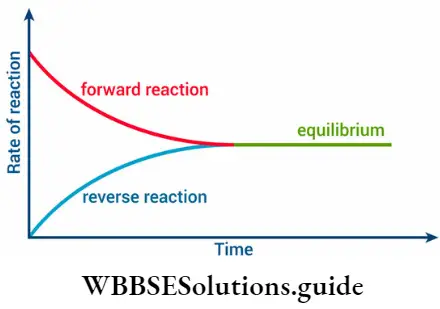
- The intensity of the colour decreases, as the reaction proceeds, and after a while, it becomes steady, indicating that an equilibrium has been reached.
- At equilibrium, it appears that the reaction has stopped, despite the presence of the reactants.
- But in reality, the rate at which hydrogen and iodine combine to form hydrogen iodide becomes equal to the rate at which hydrogen iodide decomposes.
- When we talk about the time it takes for the colour of the
reaction mixture that we have just been discussing to become
steady, we are talking about a finite time interval.
- However, to the naked eye, the change seems to be instantaneous. It is possible, though, to determine this time interval with modem instruments.
- It is also possible to establish the dynamic nature of the equilibrium using radioactive reagents.
“Physical vs chemical equilibrium multiple choice questions”
Characteristics of chemical equilibrium
1. Consider a general reversible reaction \(\mathrm{A}+\mathrm{B} \rightleftharpoons \mathrm{C}+\mathrm{D}\)
- Initially, the concentrations of A and B are maximum and the concentrations of C and D are zero. As the reaction proceeds, the concentrations of A and B decrease and the concentrations of C and D increase.
- With this, the rate of the forward reaction starts decreasing, while the rate of the backward reaction starts increasing, until the two rates become equal, and equilibrium is reached.
- At this point the concentration of A and B, and C and D become constant.
- That the concentrations of the reactants and the products become constant at equilibrium can be demonstrated by connecting a manometer to a closed vessel containing CaCO3.
If the vessel is heated, CaCO3 decomposes to yield CaO and CO2.
⇒ \(\mathrm{CaCO}_3(\mathrm{~s}) \rightleftharpoons \mathrm{CaO}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g})\)
To begin with, the pressure due to CO2 keeps increasing. Then the level of mercury in the manometer becomes steady, indicating that the pressure has become constant. This shows that the concentration of CO2 produced becomes constant (as those of CaCO3 and CaO) at equilibrium.
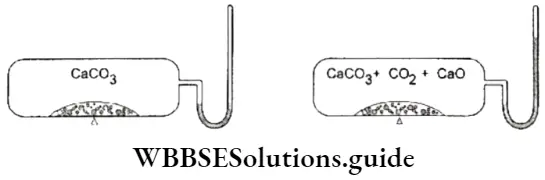
2. In the reaction that we have just considered, the pressure exerted by CO2 becomes constant at equilibrium. In the reaction between hydrogen and iodine, the colour of the reaction mixture becomes constant.
In general, the observable properties of the system become constant at equilibrium. Actually, this follows from the fact that the concentrations of the reactants and products become constant at equilibrium.
3. A chemical equilibrium can be attained only in a closed system. This should be obvious. If the system is open, one or more of the products may escape, making the reverse reaction impossible.
4. The rates of the forward and backwards reactions become equal at equilibrium. We have discussed this point at length already.
If this were not so, the reaction between hydrogen and iodine, for instance, would not appear to come to a standstill, despite the presence of the reactants.
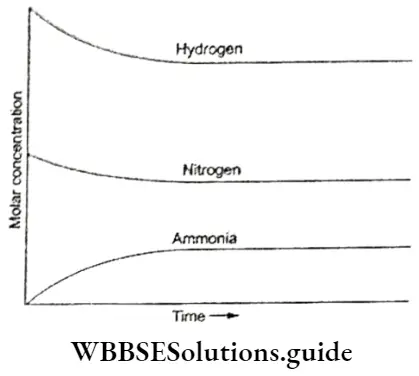
The dynamic nature of chemical equilibrium can be demonstrated in the synthesis of ammonia by the Haber process.
⇒ \(\mathrm{N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NH}_3(\mathrm{~g})\)
Known amounts of hydrogen and nitrogen are taken at high temperature and pressure, which react to form ammonia.
- The concentrations of all the three substances respectively are determined at regular intervals and a graph of the molar concentrations against time is plotted.
- The graph shows that initially the concentration of the reactants, i.c„ hydrogen and nitrogen decreases whereas that of the product ammonia increases.
- That equilibrium is reached is depicted in the graph when, after a certain time, the composition of the reaction mixture is constant.
- In order to prove the dynamic nature of equilibrium, the experiment is performed again in the same set of conditions except for the use of deuterium in place of hydrogen.
- The reaction mixture in this case, at equilibrium, has the same composition as that in the prior one except for the presence of deuterium in place of hydrogen (i.e., D2, N2 and ND3).
- Once the equilibrium is attained in both cases, the two reaction mixtures are mixed together and left. After some time the concentration of ammonia in this new mixture is found to be the same as before.
- However, mass spectrometer analysis revealed that all forms containing hydrogen as well as deuterium are present—NH3, NH2D, NHD2, ND3, H2, HD, D2—in the reaction mixture.
- This is only possible when the bonds are continuously breaking and being made at equilibrium or the forward and reverse reactions are taking place.
Had the reaction stopped then there would have been no mixing of the isotopes. This proves that the equilibrium is dynamic in nature.
5. The equilibrium can be approached from either direction. For instance, in the case of \(\mathrm{Fe}_3 \mathrm{O}_4+4 \mathrm{H}_2 \rightleftharpoons 3 \mathrm{Fe}+4 \mathrm{H}_2 \mathrm{O}\), it would make no difference whether we started with iron oxide and hydrogen, or metallic iron and steam.
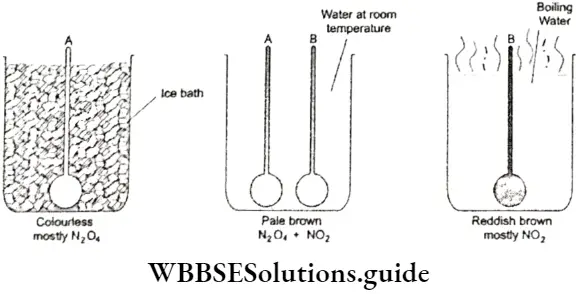
- The fact that equilibrium can be approached from either direction can be proved by performing a simple experiment. Dinitrogen tetroxide (N2O4) is a colourless gas and nitrogen dioxide (NO2) is reddish brown in colour.
- Dinitrogen tetroxide decomposes almost completely to nitrogen dioxide at 373 K. N2O4 is stable at 273 K and exists almost entirely as pure N2O4, which is colourless.
⇒ \(\underset{\text { collourless }}{\mathrm{N}_2 \mathrm{O}_4(\mathrm{~g})} \rightleftharpoons \underset{\text { reddish brown }}{2 \mathrm{NO}_2(\mathrm{~g})}\)
If you take two identical flasks A and B, fill them with nitrogen dioxide and keep them at room temperature, both will actually contain a pale brown mixture of N2O4 and NO2.
- Now place flask A in a bath maintained at 273 K and flask B in a vessel of boiling water. The gas in flask A will turn colourless, indicating that it is mostly N2O4, while the colour of the gas in flask B will deepen to reddish brown, showing that it is mostly NO2.
- If you then transfer both the flasks to a bath maintained at room temperature (298 K), the gas in flask A will turn pale brown and the reddish brown colour of flask B will fade, until both appear the same colour.
- The fact that the gaseous mixtures in both flasks attain the same colour indicates that, at equilibrium, both flasks contain a mixture of N2O4 and NO2 in the same proportion, which means that equilibrium can be approached from either direction.
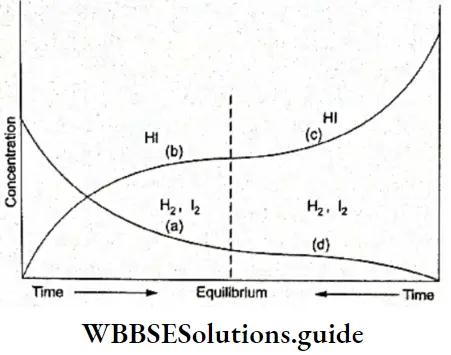
The fact is also demonstrated by Figure, which graphically represents the reaction \(\mathrm{H}_2+\mathrm{I}_2 \rightleftharpoons 2 \mathrm{HI}\) we have considered earlier in the chapter.
Whether we start the reaction from H2 and I2 or from HI, the composition of the reaction mixture at equilibrium is constant. The figure represents the forward and the backward reaction in the same graph.
- When we start the reaction with an equimolar mixture of H2 and I2, as you can see in Figure, the concentration of these two decreases (curve a) while that of HI increases (curve b). Both of them finally reach a stable concentration at equilibrium.
- When we move from right to left in the graph, we are actually starting with HI and its concentration decreases with time (curve c) while that of H2 and I2 increases (curve d), both of them stabilising at the equilibrium point.
6. You know that a catalyst increases the rate of a reaction. In the case of a reversible reaction, a catalyst increases the rates of the forward and reverse reactions to the same extent.
Hence it does not alter the state of equilibrium. That is to say, the concentrations of the reactants and products at equilibrium are the same with or without the catalyst. All that happens is that the state of equilibrium is attained faster.
7. To repeat what you already know, at equilibrium, ΔG = 0.
Law Of Mass Action
We have been discussing the fact that at equilibrium, the rates of the forward and reverse reactions become equal. But can one determine these rates? In 1863 two Norwegian chemists, C. M. Guldberg and P. Waage, came up with a law on the basis of experimental observations.
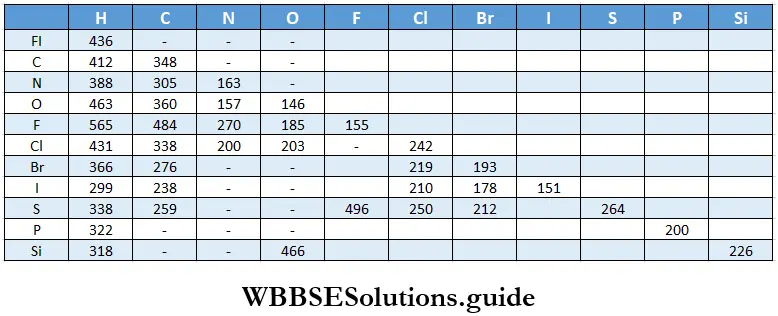
- This law, known as the law of mass action, relates the rate of chemical reaction with the concentrations of the reactants. It says that the rate at which a chemical reaction occurs at a given temperature is proportional to the product of the active masses of the reactants.
- You must remember that Guldberg and Waage came up with this law in connection with ideal gases and that the law is strictly correct only for ideal gases.
The active mass of a reactant is nothing but its molar concentration, or the number of moles dissolved per litre of the solution. For example, suppose a solution of HCl has x g of HCl dissolved in y L of solution. Then the concentration of the solution = \(\frac{x}{y} \mathrm{gL}^{-1}\)
= \(\frac{x}{36.5 \times y} \mathrm{~mol} \mathrm{~L}^{-1}\)(molar mass of HCl = 36.5)
Thus the active mass of the HCl solution, represented as [HCl], is x/36.5 y M, where M stands for molar concentration or mol L-1.
Let us consider a reaction between two reactants A and B.
A + B → products
According to the law of mass action, the rate of reaction ∝ [A] [B] or rate = k[A][B],
where [A] and [B] are the concentrations of the reactants A and B respectively, and A is a proportionality constant, called the velocity constant or rate constant.
Now consider a reaction 3C + 4D → products
you could think of this reaction as C + C + C + D + D + D + D → products then rate = k[C][C][C][D][D][D][D] = k[C]3[D]4.
In general, for any reaction aA + bB → products
∴ rate of reaction = k[A]a [B]b.
Thus, the law of mass action can also be expressed as follows.
The rate of a reaction is proportional to the product of the concentrations of the reactants, each raised to the power equal to its coefficient (number of moles of the species) in the balanced chemical equation representing the reaction.
Law Of Chemical Equilibrium
This law follows from the law of mass action. Consider the following general reversible reaction.
aA + bB \(\rightleftharpoons\)cC + dD
If we apply the law of mass action to this reaction at equilibrium, rate of forward reaction = \(k[\mathrm{~A}]_{\mathrm{eq}}^a[\mathrm{~B}]_{\mathrm{eq}}^b\),
where k is the rate constant for the forward reaction and [A]eq and [B]eq are the molar concentrations of the reactants A and B at equilibrium.
Rate of reverse reaction = \(k^{\prime}[C]_{\mathrm{eq}}^c[\mathrm{D}]_{\mathrm{eq}}^d\),
where k’ is the rate constant for the reverse reaction and [C]eq and [D]eq are the molar concentrations of the products C and D at equilibrium.
At equilibrium, rate of forward reaction = rate of reverse reaction or \(k[\mathrm{~A}]_{\mathrm{eq}}^a[\mathrm{~B}]_{\mathrm{eq}}^b=k^{\prime}[\mathrm{C}]_{\mathrm{eq}}^c[\mathrm{D}]_{\mathrm{eq}}^d\)
or, \(\frac{k}{k^{\prime}}=\frac{[\mathrm{C}]_{\mathrm{eq}}^c[\mathrm{D}]_{\mathrm{eq}}^d}{[\mathrm{~A}]_{\mathrm{eq}}^a[\mathrm{~B}]_{\mathrm{eq}}^b}=K_c\)
Generally the subscript ‘eq’ is used to denote concentration at equilibrium. The subscript ‘c’ indicates that Kc is expressed in concentrations of mol L-1.
Kc must be a constant at a given temperature since k and Ak’ are constants for a given temperature. Guldberg and Waage called it the equilibrium constant.
Thus, at equilibrium, the product of the molar concentrations of the reaction products, each raised to the power equal to its stoichiometric coefficient in the balanced chemical equation, divided by the product of the molar concentrations of the reactants, each raised to the power equal to its stoichiometric coefficient, is constant at a given temperature. This is referred to as the law of chemical equilibrium.
The equilibrium constant for the reaction \(4 \mathrm{NH}_3(\mathrm{~g})+5 \mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 4 \mathrm{NO}(\mathrm{g})+6 \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \text { is } K_{\mathrm{c}}=\frac{\left[\mathrm{NO}^4\left[\mathrm{H}_2 \mathrm{O}\right]^6\right.}{\left[\mathrm{NH}_3\right]^4\left[\mathrm{O}_2\right]^5}\)
The symbols used to denote phases in a chemical reaction are ignored while writing the expression for the equilibrium constant.
Reaction quotient: For any reversible reaction at any stage other than equilibrium, the ratio of the molar concentrations of the products to that of the reactants, where each concentration term is raised to the power equal to the stoichiometric coefficient of the substance concerned, is called the reaction quotient, Q.
It is symbolised as Qc. while molar concentrations are considered and Qp while partial pressures are considered for any reaction.
For a general reaction \(a\mathrm{~A}+b \mathrm{~B} \rightleftharpoons c \mathrm{C}+d \mathrm{D}\)
which is not at equilibrium \(Q_c=\frac{[\mathrm{C}]^c[\mathrm{D}]^d}{[\mathrm{~A}]^a[\mathrm{~B}]^b}\).
- If Qc > Kc, the value of Qc will tend to decrease to reach the value of Kc (towards equilibrium), and the reaction will proceed in the reverse direction.
- If Qc < Kc, it will tend to increase, and the reaction will proceed in the forward direction.
- If Qc = Kc, the reaction is at equilibrium.
So, if you know the relative concentrations of reactants and products at a particular stage of a reaction, you can predict the direction in which the reaction will proceed.
Also, if you know the initial concentrations of the reactants and the value of Kc, you can predict the composition of the reaction system at equilibrium.
Now consider the following reaction. \(\mathrm{CO}(\mathrm{g})+\mathrm{H}_2 \mathrm{O}(\mathrm{g}) \rightleftharpoons \mathrm{CO}_2(\mathrm{~g})+\mathrm{H}_2(\mathrm{~g})\)
Here \(Q_c=\frac{\left[\mathrm{CO}_2\right]\left[\mathrm{H}_2\right]}{[\mathrm{CO}]\left[\mathrm{H}_2 \mathrm{O}\right]}\) and Kc = 0.64 at 800°C. The initial conditions for a reaction can vary.
Here we have discussed three possibilities for the given reaction. The values taken are from different experiments. There is no need for you to memorise these values, the discussion will help you learn how to predict the direction of a reaction.
1. Only the reactants are present, say. [CO] = 0.0243 M and [H2O] = 0.0243 M.
Since the concentration of products is zero, the reaction quotient Qc = 0, i.e., Qc < Kc.
The reaction thus proceeds in the forward direction till Qc reaches a value equal to Kc.
At equilibrium, the concentrations are [CO]eq = 0.0135 M,[H2O]eq = 0.0135 M,[CO2]eq = 0.0108 M,[H2]eq = 0.0108 M.
∴ \(K_c=\frac{0.0108 \times 0.0108}{0.0135 \times 0.0135}=0.640\)
2. Only the products arc present, say. [CO2] = 0.0468 M,[H2] = 0.0468 M.
Qc >Kc and hence the reaction proceeds in the reverse direction, forming CO and H2O.
At equilibrium the concentrations are [CO]eq = 0.0260 M,[H2O]eq = 0.0260 M,[CO2]eq = 0.0208 M,[H2]eq = 0.0208 M.
∴ \(K_2=\frac{(0.0208)(0.0208)}{(0.0260)(0.0260)}\)=0.640
3. The reactants and products are present in the following concentrations.
[CO]= 0.0094 M.[HO] =0.055 M,[CO2] = 0.0005 M,[H2]= 0.0046 M.
then, \(Q_c=\frac{(0.0006)(0.0046)}{(0.0094)(0.0065)}=0.044\)
As Qc. <Kc, the reaction will proceeds in the forward direction and as expected, the concentrations at equilibrium are found to be [CO]eq = 0.0074 M; [H2O]eq = 0.0035 M; [CO2]eq =0.0025 M and [H2]eq = 0.0066 M resulting in a Kc value of 0.64.
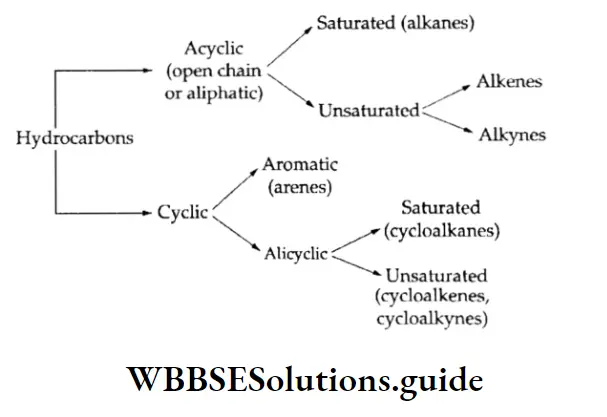
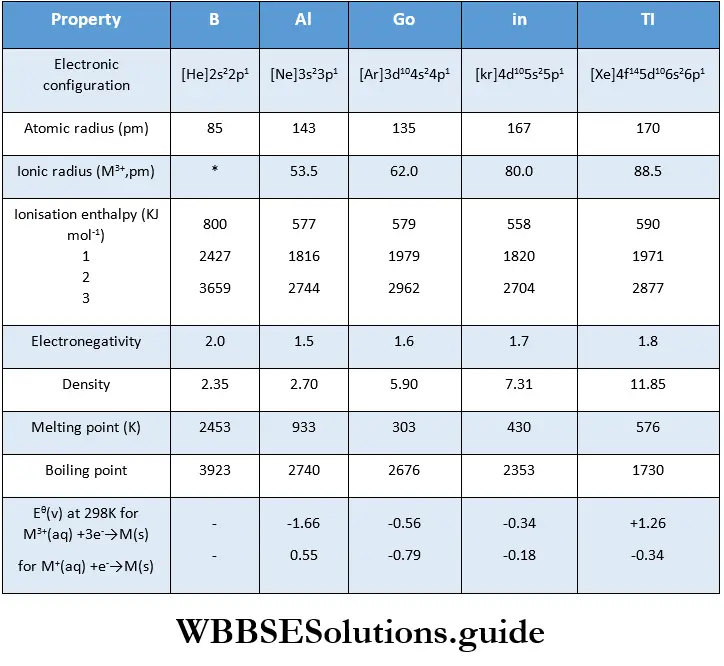
Types of equilibrium: A chemical equilibrium is called homogeneous if all the reactants and products are present in the same phase. Some examples of homogeneous equilibrium follow.
⇒ \(\mathrm{N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g})\rightleftharpoons 2 \mathrm{NH}_3(\mathrm{~g})\)
⇒ \(\mathrm{CO}(\mathrm{g})+\mathrm{H}_2 \mathrm{O}(\mathrm{g}) \rightleftharpoons \mathrm{CO}_2(\mathrm{~g})+\mathrm{H}_2\)
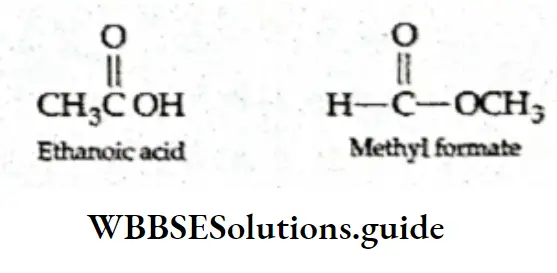
⇒ \(\mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}(\mathrm{aq}) \rightleftharpoons \mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
In a heterogeneous, chemical equilibrium, on the other hand, the reactants and products are not all in the same phases A few examples follow.
⇒ \(\mathrm{CaCO}_3(\mathrm{~s}) \rightleftharpoons \mathrm{CaO}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g})\)
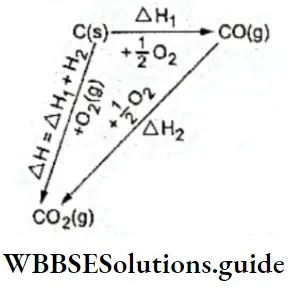
⇒ \(\mathrm{C}(\mathrm{s})+\mathrm{H}_2 \mathrm{O}(\mathrm{g}) \rightleftharpoons \mathrm{CO}(\mathrm{g})+\mathrm{H}_2(\mathrm{~g})\)
⇒ \(3 \mathrm{Fe}(\mathrm{s})+4 \mathrm{H}_2 \mathrm{O}(\mathrm{g}) \rightleftharpoons \mathrm{Fe}_3 \mathrm{O}_4(\mathrm{~s})+4 \mathrm{H}_2(\mathrm{~g})\)
∴ \(\mathrm{CrO}_4^{2-}(\mathrm{aq})+\mathrm{Pb}^{2+}(\mathrm{aq}) \rightleftharpoons \mathrm{PbCrO}_4(\mathrm{~s})\)
Gas-phase reactions: When all fee reactants and products of a reaction are gaseous, the reaction is called a gas-phase reaction and it is a case of homogeneous equilibrium.
In the case of such reactions, the equilibrium constant can be expressed either in terms of molar concentrations or partial pressures.
When expressed in terms of molar concentrations it is denoted by Kc and if it is expressed in terms of partial pressures, Kp is used. If A, B, X and Y are gases in the reaction the equilibrium constant can be expressed as follows.
⇒ \(a \mathrm{~A}+b \mathrm{~B} \rightleftharpoons x \mathrm{X}+y \mathrm{Y}\)
∴ \(K_p=\frac{\left(p_{\mathrm{X}}\right)^x\left(p_{\mathrm{Y}}\right)^y}{\left(p_{\mathrm{A}}\right)^a\left(p_{\mathrm{B}}\right)^{\mathrm{b}}}\)
In this expression, pA, pB, px and py are the partial pressures of A, B, X and Y, expressed in atmospheres or pascals.
At constant temperature, the pressure of a gas is proportional to its concentration.
In terms of concentrations, the equilibrium constant can be expressed as
∴ \(K_c=\frac{[X]^z[Y]^y}{[A]^a[B]^b} \quad \text { or } \quad \frac{\left(C_X\right)^x\left(C_Y\right)^y}{\left(C_A\right)^a\left(C_B\right)^b} \text {, }\)
where CA, CB, Cx and Cy are the molar concentrations of A, B, X and Y respectively.
If A, B, X and Y are ideal gases, pV = nRT
or \(p=\frac{n}{V} R T\)
or p =n/VRT = CRT [n/V = number of moles per litre or mol/dm3]
Then \(p_{\mathrm{A}}\)=\(\mathrm{C}_{\mathrm{A}} R T\) and \(p_{\mathrm{B}}\)=\(\mathrm{C}_{\mathrm{B}} R T\).
⇒ \(p_{\mathrm{X}}\)=\(C_{\mathrm{X}} R T\) and \(p_{\mathrm{Y}}\)=\(\mathrm{C}_{\mathrm{Y}} R T\).
∴ \(K_p=\frac{\left(C_X R T\right)^x\left(C_Y R T\right)^y}{\left(C_A R T\right)^a\left(C_B R T\right)^b}=\frac{\left(C_X\right)^x\left(C_Y\right)^y}{\left(C_{\mathrm{A}}\right)^a\left(C_B\right)^b} R T^{(x+y)-(a+b)}\).
But \(K_c=\frac{\left(C_X\right)^x\left(C_Y\right)^y}{\left(C_{\mathrm{A}}\right)^a\left(C_B\right)^b}\).
∴ \(K_p=K_c R T^{\Delta n}\),
where Δn = (x + y) – (a + b) = no. of moles of products – no. of moles of reactants in the balanced chemical equation.
Consider the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\) at equilibrium. If the equilibrium constant for the reaction is Kc then Kp would be Kp = KcRTΔn,
where Δn = no. of moles of HI- no. of moles of H2 and I2 = 2- 2 or 0.
Therefore, Kp =Kc for the reaction at equilibrium.
However, this is not always the case. Consider another example, \(\mathrm{N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NH}_3(\mathrm{~g})\) at equilibrium.
Here the number of moles of reactants is more than that of the product or Δn = 2 – 4 or -2.
Therefore, the equilibrium constant in terms of partial pressure Kp for the above reaction would be Kp = Kc(RT)-2.
If the concentration is in mol L-1 and p is in bar then R = 0.0831 L bar K-1 mol-1.
Nature of equilibrium constant
1. The equilibrium constant, as we have defined it so far, is the ratio of the product of the molar concentrations of the reaction products to that of the reactants, with each concentration term raised to the power equal to the stoichiometric coefficient of the substance in the balanced chemical equation. You may argue that if this is so, K should have units.
- Though the equilibrium constant was initially derived in terms of concentrations, strictly speaking, concentrations or partial pressures can be used only for ideal gases. However, nowadays, equilibrium constants are expressed in dimensionless quantities.
- This is possible only when the quantities (concentration and partial pressures) are measured with respect to a corresponding standard state.
- The standard state for a pure gas is taken to be 1 bar and for solute the standard state (C0) is 1 molar solution. Thus a pressure of 4 bar in terms of standard state is 4, a number without a unit.
- Therefore the numerical value of the equilibrium constant depends on the standard state chosen and both Kp and Kc are dimensionless quantities.
2. The equilibrium constant has a definite value for a particular reaction at a given temperature. This value does not depend on the initial concentrations of the reactants.
If the reaction is reversed, the tire value of K becomes the inverse of the value of the forward process. For instance, suppose the value of the equilibrium constant for the combination of hydrogen and iodine at 720 K is Kc.
⇒ \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g}) ; \quad K_c=\frac{[\mathrm{HI}]^2}{\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]} \text {. }\)
Then the value of the equilibrium constant, say K’c, for the decomposition of hydrogen iodide at the same temperature would be 1/Kc.
⇒ \(2 \mathrm{HI}(\mathrm{g}) \rightleftharpoons \mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) ; \quad K_c^{\prime}=\frac{1}{K_c}=\frac{\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]}{[\mathrm{HI}]^2}\).
3. It is all very well to say that the value of the equilibrium constant remains the same for a particular reaction at a given temperature. But a reaction may be expressed in more ways than one. For instance, the reaction between hydrogen and iodine can be expressed in the following ways.
“Homogeneous and heterogeneous equilibrium with examples”
⇒ \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\)….(1)
⇒ \(\frac{1}{2} \mathrm{H}_2(\mathrm{~g})+\frac{1}{2} \mathrm{I}_2(\mathrm{~g}) \rightleftharpoons \mathrm{HI}(\mathrm{g})\)…(2)
⇒ \(n \mathrm{H}_2(\mathrm{~g})+n \mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 n \mathrm{HI}(\mathrm{g})\) …..(3)
If the equilibrium constant for (1) is \(K_c=\frac{[\mathrm{HI}]^2}{\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]}\)
then the equilibrium constant for (2) is \(K_c^{\prime}=\frac{[\mathrm{HI}]}{\left[\mathrm{H}_2\right]^{1 / 2}\left[\mathrm{I}_2\right]^{1 / 2}}=\sqrt{K_c} \text { or } K_c^{1 / 2}\)
and the equilibrium for (3) is \(K_c^n=\frac{[\mathrm{HI}]^{2 n}}{\left[\mathrm{H}_2\right]^n\left[\mathrm{I}_2\right]^n}=K_c^n\).
Thus, the value of Kc depends on the coefficients of the reactants and products in the equation being considered.
4. If an equation is written in two steps and the values of the equilibrium constants for the two steps are K1 and K2 then the value of the equilibrium constant for the equation is the product of and K2. For example, the reaction
⇒ \(\mathrm{N}_2+2 \mathrm{O}_2 \rightleftharpoons 2 \mathrm{NO}_2 ; \quad K_c=\left[\mathrm{NO}_2\right]^2 /\left[\mathrm{N}_2\right]\left[\mathrm{O}_2\right]^2\)
occurs in two steps.
⇒ \(\mathrm{N}_2+\mathrm{O}_2 \rightleftharpoons 2 \mathrm{NO} ; \quad K_1=\left[\mathrm{NO}^2 /\left[\mathrm{N}_2\right]\left[\mathrm{O}_2\right]\right.\)
⇒ \(2 \mathrm{NO}+\mathrm{O}_2 \rightleftharpoons 2 \mathrm{NO}_2 ; \quad K_2=\left[\mathrm{NO}_2\right]^2 /\left[\mathrm{NO}^2\left[\mathrm{O}_2\right]\right.\)
∴ \(K_1K_2=\frac{[\mathrm{NO}]^2}{\left[\mathrm{~N}_2\right]\left[\mathrm{O}_2\right]} \frac{\left[\mathrm{NO}_2\right]^2}{\left[\mathrm{NO}^2\left[\mathrm{O}_2\right]\right.}=\frac{\left[\mathrm{NO}_2\right]^2}{\left[\mathrm{~N}_2\right]\left[\mathrm{O}_2\right]^2}=K_c\)
5. The value of equilibrium constant does not change if a catalyst is present.
6. The magnitude of Kc helps to predict the extent to which the reactants will be converted into the products, or the equilibrium composition of the reaction system.
- As products appear in the numerator of the equilibrium constant expression, and reactants are in the denominator, a large value of Kc or Kp indicates that the ratio of products to reactants is very large.
- In other words, the reaction proceeds nearly to completion. A very low value of Kc indicates that the reaction cannot proceed much in the forward direction.
In other words the value of Kc tells us whether the particular reaction can actually take place. Let us consider an example.
⇒ \(\mathrm{Fe}^{3+}(\mathrm{aq})+\mathrm{SCN}^{-}(\mathrm{aq}) \rightleftharpoons \mathrm{FeSCN}^{2+}(\mathrm{aq})\)
At 298 K, the equilibrium constant for the forward reaction is 138, which shows that the forward reaction is favoured or can occur. For the reverse reaction the equilibrium constant is 1/138, which shows that this reaction does not proceed much to the left.
Similarly, for the reaction, \(\mathrm{H}_2(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HCl} \text { at } 300 \mathrm{~K}\) the equilibrium constant is 4.0 x 1031. This shows that the reaction goes virtually to completion.
The equilibrium constant for the reverse reaction \(2 \mathrm{HCl}(\mathrm{g}) \rightleftharpoons \mathrm{H}_2(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g}) \text { is } \frac{1}{4.0 \times 10^{31}}=25 \times 10^{-30}\) indicating that the reverse reaction is less favoured. While a large value of Kp or Kc favours the formation of products, a very small value favours the reverse reaction or the formation of reactants.
Consider the reaction \(\mathrm{N}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}(\mathrm{g}) . \mathrm{K}_c \text { is } 4.8 \times 10^{-31}\) at 298 K for this reaction. Here, at equilibrium, the ratio of concentration of products to reactants is small, indicating that the reverse reaction is favoured.
Examples of reactions having intermediate values of equilibrium constants are
⇒ \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g}) K_c=57 \text { at } 700 \mathrm{~K}\)
⇒ \(\text { and } \mathrm{N}_2 \mathrm{O}_4(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}_2(\mathrm{~g}) K_c=4.64 \times 10^{-3} \text { at } 298 \mathrm{~K}\).
In both cases, there are appreciable amounts of reactants and products. This may be verified for the first reaction by substituting the concentrations of H2 and I2 (say 0.01 M) in the equilibrium constant expression
⇒ \(K_c=\frac{[\mathrm{HI}]^2}{\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]} \cdot\)
⇒ \({[\mathrm{HI}]^2 } =K_c \cdot\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]\)
∴ \({[\mathrm{HI}] } =\sqrt{K_c \cdot\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]}=\sqrt{57 \times 0.01 \times 0.01}=0.075 \mathrm{M}\).
It is worthwhile to mention here that the magnitude of an equilibrium constant of a reaction does not tell us anything about the rate of that reaction, i.e., how fast or how slowly the reaction will take place. It is quite possible that the equilibrium constant of a reaction may suggest completion but the rate is too low for all practical purposes.
We can make a generalisation about the extent to which a reaction proceeds based on the magnitude of Kc, as shown in Table.

Conventions regarding K
1. In the case of a homogeneous equilibrium involving products and reactants in the gaseous phase for example, \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\)
obtaining the expression for Kc or Kp is simple enough.
∴ \(K_{\mathrm{c}}=\frac{[\mathrm{HI}(\mathrm{g})]^2}{\left[\mathrm{H}_2(\mathrm{~g})\right]\left[\mathrm{I}_2(\mathrm{~g})\right]} \text { and } K_p=\frac{p_{\mathrm{HI}}^2}{p_{\mathrm{H}_2} p_{\mathrm{I}_2}} \text {. }\)
2. If one of the products or reactants involved in a heterogeneous equilibrium is a solid or a liquid its concentration is taken to be unity, by convention.
The molar concentration of a pure solid or liquid is coastant. In other words for any amount of substance ‘X’, [X(s)] and [X(l)] are constant. But [X(g)] and [X(aq)] will vary with the volume. Consider the following equilibrium.
⇒ \(\mathrm{CaCO}_3(\mathrm{~s}) \rightleftharpoons \mathrm{CaO}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g})\)
⇒ \(K_{\mathrm{c}}=\frac{[\mathrm{CaO}(\mathrm{s})]\left[\mathrm{CO}_2(\mathrm{~g})\right]}{\left[\mathrm{CaCO}_3(\mathrm{~s})\right]}=\frac{1 \times\left[\mathrm{CO}_2(\mathrm{~g})\right]}{1}=\left[\mathrm{CO}_2(\mathrm{~g})\right]\)
or \(K_p=p_{\mathrm{CO}_2}\)
This fits with the fact that the pressure of CO2 becomes constant when equilibrium is reached in the decomposition of CaCO3 in a closed vessel.
It has been found experimentally that the pressure of CO2 is 2.5 x 104 Pa when the reaction for the decomposition of CaCO3 reaches equilibrium at 1073 K. Therefore the equilibrium constant at 1073 K for the above reaction is
⇒ \(K_p=p_{\mathrm{CO}_2}=\frac{2.5 \times 10^4 \mathrm{~Pa}}{10^5 \mathrm{~Pa}}\) = 0.25
(since 1 bar =105 Pa and while calculating the value of Kp, pressure should be expressed in bar as the standard state of a substance is its pure state at exactly 1 bar.)
3. Consider an equilibrium in an aqueous medium.
⇒ \(\mathrm{NH}_3(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{NH}_4^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)
In this case H2O is the solvent and is present in a large quantity. Consequently, its concentration does not change much during the reaction and is taken to be constant (unity, by convention). In general, the concentration of a solvent is taken to be 1.
Thus, \(K_c=\frac{\left[\mathrm{NH}_4^{+}(\mathrm{aq})\right]\left[\mathrm{OH}^{-}(\mathrm{aq})\right]}{\left[\mathrm{NH}_3(\mathrm{aq})\right] \times 1}\).
4. In the light of what we have just discussed, let us reconsider the equilibrium between a liquid and its vapour in a closed system.
⇒ \(\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_2 \mathrm{O}(\mathrm{g})\)
⇒ \(K_c=\frac{\left[\mathrm{H}_2 \mathrm{O}(\mathrm{g})\right]}{\left[\mathrm{H}_2 \mathrm{O}(\mathrm{l})\right]}=\left[\mathrm{H}_2 \mathrm{O}(\mathrm{g})\right]\)
or \(K_p=p_{\mathrm{H}_2 \mathrm{O}}\).
Now do you see why the vapour pressure of a liquid is constant at a particular temperature and why it does not depend on the amount of liquid present.
5. Remember that in a chemical equilibrium the concentration of a liquid is taken as 1 only if the liquid is present as a solvent, or in a large quantity. Consider the following reaction.
⇒ \(\mathrm{CH}_3 \mathrm{COOH}(\mathrm{l})+\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}(\mathrm{l}) \rightleftharpoons \mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5(\mathrm{l})+\mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
Here \(K=\frac{\left[\mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5(\mathrm{l})\right]\left[\mathrm{H}_2 \mathrm{O}(\mathrm{l})\right]}{\left[\mathrm{CH}_3 \mathrm{COOH}(\mathrm{l})\right]\left[\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}(\mathrm{l})\right]}\)
The concentration of H2O cannot be taken as 1 in this case.
Example: A reaction between hydrogen and iodine to produce hydrogen iodide at 675 K is carried out in a closed flask of volume 4 L. At equilibrium, the reaction system contains 0.6 mol of hydrogen, 0.6 mol of iodine and 2.4 mol ofhydrogen iodide. Calculate the equilibrium constant.
Solution:
Given
A reaction between hydrogen and iodine to produce hydrogen iodide at 675 K is carried out in a closed flask of volume 4 L. At equilibrium, the reaction system contains 0.6 mol of hydrogen, 0.6 mol of iodine and 2.4 mol ofhydrogen iodide.
For the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\)
∴ \(K_{\mathrm{c}}=\frac{[\mathrm{HI}]^2}{\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]}\)
Given that,
[HI] = 2.4/4 = 0.6 mol L-1.
[H2] = 0.6/4 = 0.15 mol L-1.
[I2] = 0.6/4 = 0.15 mol L-1.
Substituting these values in the expression for Kc, \(K_c=\frac{(0.6)^2}{0.15 \times 0.15}=16\)
The equilibrium constant, the reaction quotient and Gibbs energy: As already stated, the value of the equilibrium constant for a reaction does not depend on the rate of that reaction or vice versa.
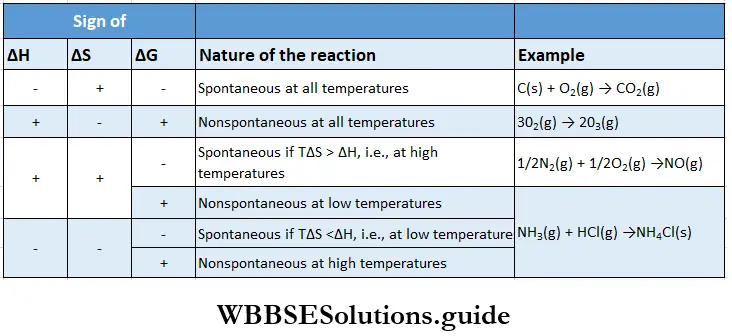
- However, it is directly related to the thermodynamics of the reaction and, in particular, to the change in free energy ΔG. You already know from the previous chapter that if ΔG is negative for a reaction then the reaction is spontaneous and should proceed in the forward direction.
- If, on the other hand, ΔG is positive, the reaction is considered nonspontaneous. Then the reverse reaction will have a negative ΔG value and will proceed, converting the products of the forward reaction into reactants.
- Either way, a reaction should proceed in the spontaneous direction until it achieves equilibrium. At this point, there is no free energy left to drive the reaction ΔG becomes zero and the reaction attains equilibrium.
This thermodynamic view of equilibrium is expressed mathematically as \(\Delta G=\Delta G^{\ominus}+R T\) ln Q
When equilibrium is achieved ΔG = 0, Q = K and this equation becomes \(\Delta G =\Delta G^{\ominus}+R T \ln K=0\)
or \(\Delta G^{\ominus} =-R T \ln K\).
Rearranging the above equation, we get
ln \(K=-\frac{\Delta G^{\ominus}}{R T}\)
or \(2.303 \log K =-\frac{\Delta G^{\ominus}}{R T}\)
or \(\log K =-\frac{\Delta G^{\ominus}}{2303 R T}\).
Taking antilog of both sides K = \(e^{-\Delta G^{\ominus} / R T}\)
or \(K=\mathrm{antilog}\left(-\frac{\Delta G^{\ominus}}{2.303 R T}\right)\)
This equation provides an interpretation of the spontaneity of a reaction.
If \(\Delta G^{\ominus}<0 \text { then } \frac{-\Delta G^{\ominus}}{R T}\) is positive, and \(e^{-\Delta C^{\ominus} / R T}>1 \text {, i.e., } K>1\). Such a reaction is spontaneous and proceeds in the forward direction so that the products are present predominantly.
Similarly, a nonspontaneous reaction is one for which \(\Delta G^\ominus>0\) and K < 1 Such a reaction proceeds in the forward direction to such a small extent that very small amounts of products are formed.
Example 1. Consider the reaction \(2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{SO}_3(\mathrm{~g})\). Amixture of SO2, SO3 and O2 at equilibrium at 1000 K has [SO2] = 3.8X10-3 M, [SO3] = 4.13X10-3 M, and [O2] = 4.3×10-3 M. Calculate Kc and Kp of the reaction.
Solution:
Consider the reaction \(2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{SO}_3(\mathrm{~g})\). Amixture of SO2, SO3 and O2 at equilibrium at 1000 K has [SO2] = 3.8X10-3 M, [SO3] = 4.13X10-3 M, and [O2] = 4.3×10-3 M.
⇒ \(K_c=\frac{\left[\mathrm{SO}_3\right]^2}{\left[\mathrm{SO}_2\right]^2\left[\mathrm{O}_2\right]}\)
Substituting the values in the expression, we get \(K_c=\frac{\left(4.13 \times 10^{-3}\right)^2}{\left(3.8 \times 10^{-3}\right)^2\left(4.3 \times 10^{-3}\right)}\)
(Note: The concentration must always be in ‘M’ or mol L-1 for calculating Kc.]
⇒ Kc = 275.
To calculate Kp we must know Δn.
Δn = no. of moles of products- no. of moles of reactants. (considering only gaseous products and reactants)
Δn = 2- (2 + 1) = 2- 3 = -1
But Kp = Kc(RT)Δn
= Kc(RT)-1
= \(\frac{K_c}{R T}\)
∴ \(K_p=\frac{275}{(0.08312)(1000)}=3.3\). [While determining the value of Kp, pressure should be expressed in bar and the value of R must be in L bar K-1 mol)-1.]
Example 2. The concentration equilibrium constant, Kc for the reaction \(2 \mathrm{NO}_2 \rightleftharpoons \mathrm{N}_2 \mathrm{O}_4\) at 25° C is 216. Find the pressure equilibrium constant, Kp, of the reaction at the same temperature.
Solution:
The concentration equilibrium constant, Kc for the reaction \(2 \mathrm{NO}_2 \rightleftharpoons \mathrm{N}_2 \mathrm{O}_4\) at 25° C is 216. Find the pressure equilibrium constant, Kp,
Kp = Kc(RT)Δn
Δn =1- 2 =-1
∴ Kp =Kc(RT)-1
= \(\frac{K_c}{R T}\)
= \(\frac{216}{(0.08312) \times(298.15)}\).
Kp = 8.72. [R = 0.0312 L bar K-1 mol-1]
Example 3. The value of Kc for the production of phosgene, the reaction being \(\mathrm{CO}(\mathrm{g})+\mathrm{Cl}_2(\mathrm{~g}) \rightleftharpoons \mathrm{COCl}_2(\mathrm{~g})\), is 5 at 600 K. What are the equilibrium partial pressures ofthe gases if the initial pressures are as follows? \(p_{\mathrm{CO}}=0.265 \mathrm{bar}, p_{\mathrm{Cl}_2}=0.265 \mathrm{bar}, p_{\mathrm{COCl}_2}=0\).
Solution:
The value of Kc for the production of phosgene, the reaction being \(\mathrm{CO}(\mathrm{g})+\mathrm{Cl}_2(\mathrm{~g}) \rightleftharpoons \mathrm{COCl}_2(\mathrm{~g})\), is 5 at 600 K.
At first, Kp must be calculated from Kc.
Kp = Kc(RT)Δn
Here, Δn =1- 2 = -1
∴ Kp = 5 x (0.08312 x 600)-1 = 0.1
Let us now tabulate the partial pressures of all the gases. Suppose the partial pressure of COCl2 at equilibrium is x bar.
Partial pressures:
⇒ \(\begin{array}{lccc}p_{\mathrm{CO}} p_{\mathrm{CO}_2} p_{\mathrm{COCl}_2} \\
\text { Initial } 0.265 \text { bar } 0.265 \text { bar } 0\end{array}\)
⇒ \(\begin{array}{lccc}
\text { Change } -x -x -x \\
\text { Equilibrium } 0.265-x 0.265-x x
\end{array}\)
∴ \(\dot{K}_p=0.1=\frac{p_{\mathrm{COC}_2}}{p_{\mathrm{CO} p_{\mathrm{Cl}_2}}}=\frac{x}{(0.265-x)^2}\)
0.1 = \(\frac{x}{\left(0.07-0.53 x+x^2\right)}\)
Rearranging, we get
0.1 x2 – 0.053x + 0.007 = x
or 0.1 x2 – 0.053x – x + 0.007 = 0
or 0.1 x2 – 0.053 x + 0.007 = 0
or x = \(\frac{+1.053 \pm \sqrt{(1053)^2-4 \times 0.1 \times 0.007}}{2 \times 0.1}=\frac{1.053 \pm 1.0517}{0.2}=6.5 \times 10^{-3} \text { or } 10.52\)
x cannot be greater than 0.265 bar since even if all of CO and Cl2 react, \(\mathrm{p}_{\mathrm{COCl}_2}\), cannot be more than 0.265 bar.
Choosing x to be 6.5 x 10-3
at equilibrium, \(\mathrm{p}_{\mathrm{COCl}_2}\) =6.5 x 10-3 bar
PCO = 0-265 – 65 x 10-3 = 0.2585 bar = pa
Example 4. The standard Gibbs free energy at 298 K for the reactions are given.
- \(\mathrm{N}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}(\mathrm{g}) \Delta \mathrm{G}^{\ominus}=173.2 \mathrm{~kJ}\)
- \(2 \mathrm{NO}(\mathrm{g})+\mathrm{O}_2(\mathrm{~g}) [latex] \rightleftharpoons 2 \mathrm{NO}_2(\mathrm{~g}) \Delta \mathrm{G}^{\ominus}=-69.7 \mathrm{~kJ}\)
Calculate Kp at 298 K for the following reaction \(\mathrm{N}_2(\mathrm{~g})+2 \mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}_2(\mathrm{~g})\)
Solution:
First, we must find \(\Delta G^{\ominus}\), for the reaction of interest.
⇒ \(\begin{aligned}
\mathrm{N}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}(\mathrm{g}) \quad \Delta \mathrm{G}^{\ominus}=173.2 \mathrm{~kJ} \\
2 \mathrm{NO}(\mathrm{g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}_2(\mathrm{~g}) \quad \Delta G^{\ominus}=-69.7 \mathrm{~kJ} \\
\overline{\mathrm{N}_2(\mathrm{~g})+2 \mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}_2(\mathrm{~g}) \Delta G^{\ominus}=-173.2-69.7} \\
=0.103 .5 \mathrm{~kJ} \text {. } \\
\end{aligned}\)
Thus, \(\Delta G^{\ominus}\) for the reaction in question is the sum of \(\Delta G^{\ominus}\) for the other two reactions.
∴ \(\Delta G^{\ominus}\) = 103.5 kJ.
∴ \(K_p=e^{-\Delta G^{\ominus} / R T}\)
But \(\frac{-\Delta G^{\ominus}}{R T}=\frac{-103.5 \times 1000}{8.314 \times 298}=-4178\)
∴ \(\quad K_p=e^{-41.78}=7.16 \times 10^{-19}\).
Example 5. \(\Delta_f G^{\ominus}\) for ammonia gas is -16.5 kJ mol1 at 298 K. Find the equilibrium constant for the reaction \(\mathrm{N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g}) \rightarrow 2 \mathrm{NH}_3(\mathrm{~g})\)
Solution:
2 mol of NH3 is produced in the reaction but the \(\Delta_f G^{\ominus}\) value given is for only 1 mol. \(\Delta_f G^{\ominus}\) for the formation of 2 mol of NH3 is equal to twice \(\Delta_f G^{\ominus}\)for 1 mol.
⇒ \(\Delta_f G^{\ominus}\) = 2 x -165 kJ = -33.0 kJ = -33,000 J.
Now, \(\frac{-\Delta_f G^{\ominus}}{R T}=\frac{33000}{8.314 \times 298}=13.32\)
∴ \(K_c=e^{-\Delta, G^4 / R T}=e^{+1332}=609,259.8=6.09 \times 10^5\).
Example 6. Kc for the hydrolysis of sucrose is 5.3 x 1012 at 298 K. Find \(\Delta G^{\ominus}\) for the process sucrose + H2O \(\rightleftharpoons\) glucose + fructose
Solution:
Given
Kc for the hydrolysis of sucrose is 5.3 x 1012 at 298 K.
⇒ \(K_c=e^{-\Delta G^{\ominus} / R T}\)
ln \(K_c=-\Delta G^{\ominus} / R T\)
⇒ \(\Delta G^{\ominus}=-R T \ln K\)
= -8.314 x 298 x ln(53 x 1012) = -72,589.7 J.
∴ \(\Delta G^{\ominus}\)=-72.58 kJ.
Example 7. Consider the reaction \(\mathrm{PCl}_5(\mathrm{~g}) \rightleftharpoons \mathrm{PCl}_3(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g})\). Kp =23.6 at 500K for the reaction. Calculate the equilibrium partial pressures of the reactants and products if the initial pressure of PCl5 is 0.56 bar and those of PCl3 and Cl2 are zero. How will the concentration of PCl5 and PCl3 change if more Cl2 is added once equilibrium is reached?
Solution:
Given
Consider the reaction \(\mathrm{PCl}_5(\mathrm{~g}) \rightleftharpoons \mathrm{PCl}_3(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g})\). Kp =23.6 at 500K for the reaction.
Let us tabulate the partial pressure—initial, change and at equilibrium.
⇒ \(p_{\mathrm{PC}} p_{\mathrm{PC}_3} p_{\mathrm{C}_2}\)
⇒ \(\begin{array}{lccc}
\text { Initial } 0.56 0 0 \\
\text { Change } -x +x +x \\
\text { Equilibrium } 0.56-x x x
\end{array}\)
Putting these values in the expression for Kp,
⇒ \(K_p=\frac{p_{\mathrm{PCl}_3} p_{\mathrm{Cl}_2}}{p_{\mathrm{PCl}_5}}\)
23.6 = \(\frac{x \cdot x}{0.56-x}\)
Rearranging, we get 23.6(056- x)- x2 = 0
or x2– 23.6(056 -x) = 0
or x2 23.6 x 056 + 23.6x = 0
or x2+23.6x- 13.2 = 0.
∴ x = \(\frac{-23.6 \pm \sqrt{(23.6)^2+4 \times 13.2}}{2}\)
= \(\frac{-23.6 \pm 24.69}{2}=0.547 \text { or }-24.14\)
Taking the positive root, x = 0547, we get
⇒ \(p_{\mathrm{PCl}_5}\) = 0.56 -0547 = 0.013 bar
⇒ \(p_{\mathrm{PCl}_3}\) = 0.547 bar
⇒ \(p_{\mathrm{Cl}_2}\) = 0547 bar,
On adding Cl2 at equilibrium, the reaction will shift towards the left \(p_{\mathrm{PCl}_5}\) and will increase and \(p_{\mathrm{PCl}_3}\) will decrease.
Factors Affecting Equilibrium
In this section, we shall discuss what happens to a system at equilibrium when certain conditions are changed. We shall consider various changes, one by one.
- What happens to the equilibrium when the temperature is changed, for example? Or what happens when the concentration of either the reactants or the products is changed?
- In 1888, the French chemist Henri Louis Le Chatelier proposed a general law on the behaviour of a system in equilibrium.
- This law, called Le Chatelier’s principle, states that if a system at equilibrium is subjected to a change which displaces if from the equilibrium, a net reaction will occur in a direction that counteracts the change.
- This law is applicable to all physical and chemical equilibria. Let us now discuss in detail the various factors that can affect an equilibrium.
Change in concentration: Let us consider the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\) at equilibrium.
- If more of the reactant H2 or I2, or both, are added to the reaction system, the state of equilibrium will be disturbed and the tendency of the system would be to counteract this change so that equilibrium is established again.
- The only way this can happen is if more of the reactants react to form more of the products so that the ratio of the products of the concentrations the reaction products to that of the reactants, i.e., K, remains constant.
- Another way of saying this is, if the concentration of the reactants is increased, the equilibrium shifts in the forward direction, or the forward reaction is favoured.
- Applying Le Chatelier’s principle to a specific change (concentration), we may say, “When the concentration of any of the reactants or products in a reaction at equilibrium is changed, the composition of the equilibrium mixture changes so as to undo the effect of the concentration change.”
Let us study how equilibrium is re-established if we add H2 at equilibrium. The concentration of H2 increases and equilibrium is disturbed.
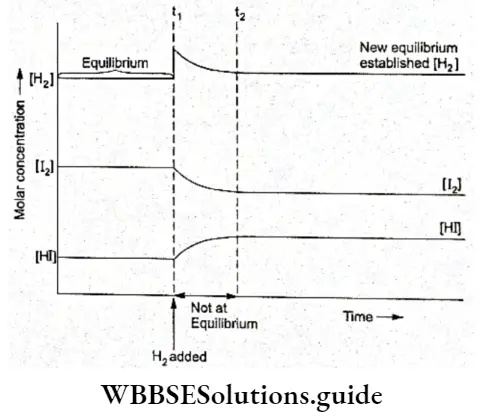
A new equilibrium will be set up in which the concentration of H2 should be less than what it is after adding H2. But the initial concentration of H2 increases for the new equilibrium as shown in Figure.
- As you can see from the figure, till the time tx, the system is at equilibrium with the concentrations of H2, I2 and HI being given by the intercept of the three curves on the y-axis.
- At t1, when some H2 is added, its concentration increases as shown by the steep rise of the H2 curve. The equilibrium is disturbed and the system responds to the change by forming more HI, thus decreasing the concentration of H2 and I2 than at time t1. At t2 a new equilibrium is established.
“Le Chatelier’s principle and factors affecting equilibrium”
Let us consider the reaction quotient for this reaction \(Q_c=\frac{[\mathrm{HI}]^2}{\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]}\)
If hydrogen is added at equilibrium, the molar concentration of hydrogen increases and the equilibrium is disturbed. The denominator of the above equation increases and hence Qc decreases or becomes less than Kc.
- Therefore, in order to make Qc equal to Kc the numerator must increase or the denominator must decrease. This means more HI should be formed in order to counteract the disturbance in equilibrium.
- As a result more of H2 and I2 react to form more of HI to re-establish the equilibrium making the forward reaction more favourable.
- What if the concentration of hydrogen iodide is increased? Again there will be a tendency for the reaction system to counteract this change.
- This time, more hydrogen iodide will yield more hydrogen and iodine so that equilibrium is restored and Kc remains constant. That is to say, if the concentration of the products is increased, the equilibrium will shift in the reverse direction, or the reverse reaction will be favoured.
Changes in concentration (of either products or reactants) play an important role in the productivity of industrial processes. When ammonia is manufactured by the Haber process, for example, the equilibrium involved is the following.
⇒ \(3 \mathrm{H}_2(\mathrm{~g})+\mathrm{N}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NH}_3(\mathrm{~g})\)
The product, i.e., NH3, is continuously removed from the site of the reaction. This disturbs the equilibrium and the system counteracts the change by producing more ammonia.
- Increasing the concentration of the reactants would have the same impact. But, obviously, the former is more economical.
- The large-scale production of CaO from CaCO3 is another example: the constant removal of CO2 from the lime kiln drives the reaction to completion.
- There are many applications of Le Chatelier’s principle in our everyday lives. For instance, on humid, cloudy days when there is hardly any breeze, we often dry clothes indoors, under the fan.
- When we do this, we are using Le Chatelier’s principle without knowing it. The water present in the clothes and the water vapour in the air in the vicinity of the clothes reach an equilibrium, which we disturb by turning on the fan.
- The artificial breeze (created by the fan) removes the water vapour from the immediate neighbourhood of the clothes and more water molecules from the clothes escape into the air to re-establish equilibrium.
- For the same reason, one feels more comfortable under the fan on a sultry day.
- A very vital life process depends on Le Chatelier’s principle. You know that haemoglobin (ITb) present in red blood corpuscles acts as a carrier of oxygen from the lungs to the tissues and as a carrier of carbon dioxide from the tissues to the lungs.
How does this happen? The blood that comes from the lungs and reaches the tissues has a high concentration of oxygen in comparison with the blood present in the tissues, where the partial pressure of oxygen is low.
This disturbs the equilibrium and in order to re-establish it, some of the oxyhaemoglobin (haemoglobin carrying oxygen) dissociates.
⇒ \(\mathrm{HbO}_2(\mathrm{~s}) \rightleftharpoons \mathrm{Hb}(\mathrm{s})+\mathrm{O}_2(\mathrm{~g})\)
When the blood returns from the tissues to the lungs, more oxyhaemoglobin is formed.
- This happens because the concentration or partial pressure of oxygen in the lungs is high and to re-establish equilibrium, some of the haemoglobin in the blood returning from the tissues combines with oxygen to form oxyhaemoglobin.
- The removal of carbon dioxide from the tissues by haemoglobin happens in a similar manner.
- The partial pressure of carbon dioxide in the tissues is high, so some of it dissolves in the blood, which reaches the tissues from the lungs.
Carbon dioxide is released from the blood when it returns to the lungs, where the partial pressure of carbon dioxide is low.
⇒ \(\mathrm{CO}_2(\mathrm{~g})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_2 \mathrm{CO}_3(\mathrm{aq}) \rightleftharpoons \mathrm{H}^{+}(\mathrm{aq})+\mathrm{HCO}_3^{-}(\mathrm{aq})\)
The effect of concentration can also be demonstrated with the help of a simple experiment. You are familiar with the reaction
⇒ \(\underset{\text { yellow }}{\mathrm{Fe}^{3+}(\mathrm{aq})}+\underset{\text { colourless }}{\mathrm{SCN}^{-}(\mathrm{aq})} \rightleftharpoons \underset{\text { deep red }}{\mathrm{FeSCN}^{2+}(\mathrm{aq})}\)
⇒ \(K_c=\frac{\left[\mathrm{FeSCN}^{2+}(\mathrm{aq})\right]}{\left[\mathrm{Fe}^{3+}(\mathrm{aq})\right]\left[\mathrm{SCN}^{-}(\mathrm{aq})\right]}\)
If we take ferric nitrate solution and thiocyanate in a test tube in an appropriate proportion and shake, the colour changes from yellow to red due to the formation of FeSCN2+.
- This happens gradually and the colour does not change once equilibrium is attained. We can shift the equilibrium by adding or removing the reactant or product.
- If we add few more drops of thiocyanate solution in the test tube, Qc becomes less than Kc. Therefore, the forward reaction is favoured and more FeSCN2+ is formed until Qc = Kc and equilibrium is attained.
- At first, the intensity of the red colour decreases and then increases to deep red, at equilibrium.
- If on the other hand, we remove SCN– by adding a reagent, say HgCl2(aq) which forms a stable complex—[Hg(SCN)4]2-—the equilibrium shifts in the reverse direction so that the concentration of FeSCN2+( decreases because it dissociates to replenish SCN–.
- A similar shift in the equilibrium will be observed if we add oxalic acid to the reaction mixture at equilibrium. Oxalic add reacts with Fe3+ to form a stable complex—[Fe(C2O4)3]3-.
Change in pressure: If one of the reactants or products in a reaction system is in the gaseous phase, pressure becomes an important factor governing shifts in equilibrium. In the case of a gas, increase in partial pressure amounts to increase in concentration.
- Let us consider a heterogeneous system first. \(\mathrm{CO}_2(\mathrm{~g})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{CO}_2(\mathrm{aq})\)
- In this case increasing the partial pressure of CO2(g) would mean an increase in the number of moles of CO2 per unit volume (concentration).
- According to Le Chatelier’s principle the system would try to counteract the change to re-establish equilibrium. The only way this is possible is if more CO2 dissolves in water. This is why the solubility’ of gases increases with increase in pressure.
- Now consider a homogeneous system in which all the reactants and products are in the gaseous phase. The reaction could lead to an overall increase in the number of moles, if the number of moles of the products is more than that of the reactants.
It could lead to an overall decrease in the number of moles, if the number of moles of products is less than that of the reactants. There is a third possibility—there may be no net change in the number of moles. Let us consider these cases one by one.
1. The reaction involved in the manufacture of ammonia by the Haber process is as follows. \(3 \mathrm{H}_2(\mathrm{~g})+\mathrm{N}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NH}_3(\mathrm{~g})\)
- This is obviously a case in which the forward reaction leads to a decrease in the number of moles.
- In such a case, if the total pressure is increased, say by decreasing the volume, the number of moles per unit volume increases and the system tries to nullify the change by decreasing the number of moles per unit volume (we already know that pV = constant and p ∝ moles of the gas).
- Since the forward reaction leads to a decrease in the number of moles, it is favoured and the equilibrium shifts in the forward direction.
- In short, increasing the pressure leads to an increase in the yield of the products. Obviously, if pressure is decreased, the reverse reaction will be favoured.
This can also be understood by considering reaction quotient. Let[H2], [N2] and [NH3]be the molar concentrations at equilibrium in the reaction involved in the Haber process.
When the volume of the reaction mixture is halved the pressure as well as concentrations are doubled. Now, let us obtain the concentration quotient by replacing each value by its double.
⇒ \(Q_c=\frac{\left(2\left[\mathrm{NH}_3\right]\right)^2}{\left(2\left[\mathrm{H}_2\right]\right)^3\left(2\left[\mathrm{~N}_2\right]\right)}=\frac{1}{4} \frac{\left[\mathrm{NH}_3\right]}{\left[\mathrm{H}_2\right]\left[\mathrm{N}_2\right]}=\frac{K_c}{4}\)
As Qc < Kc, the forward reaction is favoured.
2. Let us consider an equilibrium in which the forward reaction involves an increase in the number of moles.
⇒ \(\mathrm{PCl}_5(\mathrm{~g}) \rightleftharpoons \mathrm{PCl}_3(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g})\)
Here an increase in pressure will favour the backward reaction, while a decrease in pressure will favour the forward reaction.
3. In certain reactions, for example, the ones which follow, the number of moles of the products is equal to that of the reactants.
⇒ \(\mathrm{N}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NO}(\mathrm{g})\)
⇒ \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\)
Changes in pressure have no impact on such reactions.
When we study the effect of change of pressure on a heterogenous system, the solid or liquid reactants or products are ignored because the volume (and concentration) of a solid or a liquid is nearly independent of pressure.
For instance, in the following reactions carbon and iodine are solids.
⇒ \(\mathrm{C}(\mathrm{s})+\mathrm{CO}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{CO}(\mathrm{g})\)
⇒ \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~s} \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\)
When the pressure on the reaction mixture is increased, the reverse reaction is favoured because the number of moles of the gases decreases in the reverse direction.
Change in temperature: Temperature affects the equilibrium constant Kc. The nature and extent of the change in Kc or Kp of a reaction due to the change in temperature depends on the enthalpy change of the reaction.
Suppose the forward reaction in a chemical equilibrium is exothermic then the reverse reaction will, naturally, be endothermic. Production of ammonia by the Haber process involves an exothermic reaction.
⇒ \(\mathrm{N}_2(\mathrm{~g})+3 \mathrm{H}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{NH}_3(\mathrm{~g}) ; \Delta_r H^{\ominus}=-9238 \mathrm{~kJ} \mathrm{~mol}^{-1}\)
- If such a reaction system attains equilibrium at a particular temperature and then heat is supplied to the system so that the temperature increases, the equilibrium gets disturbed.
- According to Le Chatelier’s principle, the system then tries to counteract the change in temperature and the only way it can do this is by absorbing heat.
- In other words, the equilibrium shifts to the left and the endothermic or reverse reaction is favoured.
- If, on the other hand, the temperature is decreased, the equilibrium shifts forward, favouring the forward or exothermic reaction, thus leading to high yield of ammonia.
In this context, refer to the experiment in Figure. NO2 dimerises into N2O4 according to the following reaction.
⇒ \(2 \mathrm{NO}_2(\mathrm{~g}) \rightleftharpoons \mathrm{N}_2 \mathrm{O}_4(\mathrm{~g}) ; \Delta_r H^{\ominus}=-57.2 \mathrm{~kJ} \mathrm{~mol}^{-1} \text {. }\)
Here the formation of N2O4 is favoured at a low temperature (ice bath) whereas that of NO2 is favoured at high temperature (hot water).
This reaction is exothermic. On increasing the temperature, in order to relieve the stress of added heat, the system will tend to absorb this heat and the reverse reaction will be favoured.
- Since NO2 is brown and N2O4 is colourless, the effect of increasing temperature on this reaction may easily be seen.
- Decreasing the temperature, say by immersing the vessel containing a mixture of NO2 and N2O4 in an ice bath, favours the forward reaction.
- When the dissolution of a solid in water (or any other solvent) is accompanied by the absorption (endothermic) or release (exothermic) of heat, its solubility changes with temperature.
- When NH4Cl dissolves in water, for example, heat is absorbed. The solubility of NH4Cl increases with temperature because an increase in temperature favours the endothermic process.
- CaCl2, on the other hand, dissolves in water with the evolution of heat. The solubility of CaCl2, thus, decreases with increase in temperature.
The solubility of NaCl is almost unaffected by changes in temperature because there is very little heat change during the dissolution of NaCl.
Effect of a catalyst: A catalyst merely changes the time taken to reach the state of equilibrium, it does not change the equilibrium constant. It lowers the activation energy of the forward and reverse reactions by the same amount.
- The activation energy is the energy barrier that has to be overcome for the reaction to proceed. If a reaction mixture is not at equilibrium, a catalyst accelerates the rate at which equilibrium is reached but it does not affect the composition of the equilibrium mixture.
- Hence it does not appear in the balanced chemical equation or in the equilibrium constant expression.
- It increases the rates of the forward and reverse reactions to the same extent. Nonetheless, the fact that a catalyst decreases the time taken to reach equilibrium is very important for many industrial processes.
- For instance, iron-molybdenum is used as a catalyst in the production of ammonia from nitrogen and hydrogen because at low temperature, the rate of the reaction is very slow and at high temperature, the reverse reaction is favoured.
- The optimum conditions of temperature and pressure for the synthesis of NH3 using a catalyst are 400-500°C and 130-300 atm.
In the manufacture of sulphuric acid by the contact process, the conversion of SO2 to SO3 is very important.
⇒ \(2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{SO}_3(\mathrm{~g}) ; K_{\mathrm{c}}=1.7 \times 10^{26}\)
The magnitude of the equilibrium constant indicates that the reaction should reach completion.
However, the oxidation of SO2 is very slow but in the presence of the catalyst V2O5 (vanadium pentoxide), the rate of the reaction increases. But if a reaction has a very low Kc, a catalyst would be of no help.
Effect of inert gas: While studying the effect of pressure, we have considered changes that result from a change in volume (as volume and pressure are inversely proportional).
- But what if we keep the volume constant and increase the pressure by adding an inert gas. The inert gas is added just to change the pressure—it does not take part in the reaction.
- We could also think of adding an inert gas at constant pressure. If an inert gas is added to a reaction system containing reactants and products in the gaseous phase at equilibrium, the effect it will have on the equilibrium will depend on whether it is added at constant volume or at constant pressure.
- If the equilibrium is readied at constant volume, i.e., in a closed vessel, the addition of the inert gas will not change the molar concentrations (or partial pressures) of the reactants and products. Consequently, the state of equilibrium will not be affected.
If an inert gas is added to a system at equilibrium at constant pressure, the volume will increase.
- An increase in volume will lead to a decrease in the molar concentrations (or partial pressures) of all the reactants and products.
- If the reaction is such that the number of moles of the reactants is the same as that of the products, a decrease in the molar concentrations of all the reactants and products will make no difference to the equilibrium constant since it is the ratio of the product of concentrations of the products to that of the reactants.
- (We came across a similar situation while studying the effect of a change in pressure on an equilibrium.) If, however, the number of moles of the reactants is not the same as that of the products, an increase in the total volume will disturb the equilibrium.
Suppose the number of moles of tire reactants is less than that of the products, as in the following equilibrium.
⇒ \(\mathrm{PCl}_5(\mathrm{~g}) \rightleftharpoons \mathrm{PCl}_3(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g})\)
In such a case, a decrease in the molar concentrations (due to increase in total volume) of all the products and reactants will lead the system to counteract the change and the equilibrium will shift in the direction of increasing the number of moles, i.e., the forward direction.
- We can look at it in another way. At equilibrium \(K_{\mathrm{c}}=\frac{\left[\mathrm{PCl}_3\right]\left[\mathrm{Cl}_2\right]}{\left[\mathrm{PCl}_5\right]}\)
- If the concentrations of all the reactants and products decrease, the numerator will decrease more since there are two concentration terms in the numerator and Kc will decrease.
- For Kc to remain constant, the equilibrium will shift in the direction of an increase in the concentrations of products and decrease in the concentration of the reactants, i.e., in the forward direction.
Similarly, if a reaction is such that the number of moles of the reactants is more than that of the products, an increase in volume will push the equilibrium in the reverse direction.
⇒ \(2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{SO}_3(\mathrm{~g})\)
“Applications of equilibrium in real life chemistry”
For example, for the reaction an increase in volume will favour the reverse reaction. In general, the addition of an inert gas at constant pressure (to a system at equilibrium) makes the equilibrium shift in the direction that increases the number of moles.
Ionic Equilibrium
When certain compounds like NaCl, KCl or NaOH are dissolved in water, the resultant solution is a good conductor of electricity. The solution of NaCl contains Na+ and Cl– ions surrounded by water.
⇒ \(\mathrm{NaCl}(\mathrm{s}) \stackrel{\mathrm{H}_2 \mathrm{O}}{\longrightarrow} \mathrm{Na}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
⇒ \(\mathrm{KCl}(\mathrm{s}) \stackrel{\mathrm{H}_2 \mathrm{O}}{\longrightarrow} \mathrm{K}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
- The electrostatic attraction between the ion and water (ion-dipole interaction) results in the dissolution of such ionic compounds in water. The figure shows various interactions in the solution of KCI in water.
- When KCl is dissolved in water, the positive end of the polar water molecule (hydrogen) is attracted by negative chloride ions at the surface of the solid, similarly, the negative end of the water molecule (oxygen) is attracted by the positive potassium (K+) ions.
- The water molecules penetrate between K+ and Cl– and surround them, thus reducing the strong interionic forces holding them together and allowing them to move as hydrated ions.
- This process is called ionisation, i.e., the process of breaking up of a compound into its ions in a solution.
- Though the exact definition of ionisation differs from that of dissociation, we have used both the terms to refer to the breaking up of an ionic substance into its constituent into ions for the sake of convenience.
- Michael Faraday in 1824 classified substances into electrolytes and nonelectrolytes. Nonelectrolytes do not conduct electricity.
- Electrical conductivity requires the existence of charged particles and in a solution of nonelectrolyte, the solute molecules retain their unbroken identity.
That is why though sucrose dissolves in water, it is not a conductor of electricity.
⇒ \(\mathrm{C}_{12} \mathrm{H}_{22} \mathrm{O}_{11}(\mathrm{~s}) \stackrel{\mathrm{H}_2 \mathrm{O}}{\longrightarrow} \mathrm{C}_{12} \mathrm{H}_{22} \mathrm{O}_{11}(\mathrm{aq})\)
Not all electrolytes conduct electricity with equal ease. Faraday categorised electrolytes as strong and weak. For instance, sodium chloride is a strong electrolyte whereas acetic acid is weak electrolyte.
Ionisation: All electrolytes do not ionise to the same extent. The extent to which an electrolyte ionises is called its degree of dissociation.
- More accurately, the fraction of the total number of molecules of the electrolyte in the solution which dissociates into ions is called the degree of dissociation of the electrolyte.
- A truly ionic compound dissociates completely in solution. For example, if 1 mol of NaCl is dissolved in 1 L of water, the solution will contain 1 mol of Na+ and 1 mol of Cl– ions.
- Compounds which dissociate completely in solution are called strong electrolytes.
- Polar covalent compounds may also produce ionic solutions or solutions which conduct electricity.
- Depending on the degree of ionisation or dissociation, such solutes are strong electrolytes or weak electrolytes.
H2SO4, for example, dissociates completely in solution and is a strong electrolyte. Equations for the ionisation of strong electrolytes are written with a single arrow.
⇒ \(\mathrm{HCl}(\mathrm{aq}) \longrightarrow \mathrm{H}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
⇒ \(\mathrm{NaOH}(\mathrm{aq}) \longrightarrow \mathrm{Na}^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)
Weak electrolytes, like NH3 and CH3COOH (acetic acid), on the other hand, do not dissociate completely. Their solutions arc weakly or partly ionised.
In such cases there is an equilibrium between the un-ionised molecules of the electrolyte and the ions in solution. Consequently, the ionisation of such an electrolyte is represented with a double arrow.
⇒ \(\mathrm{NH}_3(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{NH}_4^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)
⇒ \(\mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})\)
- The degree of ionisation of weak electrolytes depends upon the nature of the electrolyte, temperature and dilution.
- We will not concern ourselves with the ionisation of strong electrolytes because such reactions proceed to completion and in this chapter we are examining reactions from the point of view of equilibrium.
Ionisation of weak electrolytes: An equilibrium between the (un-ionised) molecules of a weak electrolyte and its ions in solution is called an ionic equilibrium.
- If C is the concentration of an electrolyte in a solution, and a is the degree of dissociation, the number of moles of the electrolyte that dissociate is Cα.
- The degree of dissociation (α) of an electrolyte is the fraction of the total number of molecules of the electrolyte (in the solution) that ionise at equilibrium.
- For example, if the value of α = 0.5 or 1/2, it means that out of 1 mole of the electrolyte, 0.5 mol has ionised or there is 50% ionisation.
- So, if C is the concentration of the electrolyte, what is the concentration that is ionised? It is Cα.
Let us say that the concentration of some electrolyte XY in an aqueous solution is C and its degree of dissociation is a. At equilibrium, the ionisation of the electrolyte can be represented by
⇒ \(\mathrm{XY}(\mathrm{aq}) \rightleftharpoons \mathrm{X}^{+}(\mathrm{aq})+\mathrm{Y}^{-}(\mathrm{aq})\)
At equilibrium, the concentrations of X+ and Y– must each be Cα. And the concentration of XY must be C -Cα = C(1 – α). (The concentration of X+ and Y– are equal to the concentration of XY dissociated.)
Applying the law of chemical equilibrium
K = \(\frac{\left[\mathrm{X}^{+}(\mathrm{aq})\right]\left[\mathrm{Y}^{-}(\mathrm{aq})\right]}{[\mathrm{XY}(\mathrm{aq})]}\)
= \(\frac{(\mathrm{C} \alpha)(\mathrm{C} \alpha)}{C(1-\alpha)}=\frac{C \alpha^2}{1-\alpha}\).
For a weak electrolyte, a is so small compared to 1 that it can be neglected in approximate calculations. Then K = Cα2
or \(\quad \alpha^2=\frac{K}{C}\)
or \(\alpha=\sqrt{\frac{K}{C}}\)
⇒ \(\quad \alpha \propto \sqrt{\frac{1}{C}}\) (because K is a constant)
- Therefore, for a weak electrolyte, the degree of ionisation is inversely proportional to the square root of the molar concentration.
- This is known as Ostwald’s dilution law. As C approaches zero, or dilution approaches infinity, the degree of dissociation approaches unity.
- This means that the ionisation of a weak electrolyte will be more in a dilute solution than in a concentrated solution.
Example: Calculate the degree of dissociation of a 0.02-M solution of formic acid, given that K = 1.9 x 10-4.
Solution:
The dissociation of formic acid in water can be represented as follows.
⇒ \(\mathrm{HCOOH}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{HCOO}^{-}\)
Let α be the degree of dissociation. Then the concentrations of the ionic species at equilibrium will be as follows:
[H3O]+ = 0.02α
[HCOO]– = 0.02α
[HCOOH] = 0.02 (1 – α) = 0.02, assuming a to be negligible compared to 1.
Now K = \(\frac{(0.02 \alpha)(0.02) \alpha}{0.02}=1.9 \times 10^{-4}\) (given)
or \(\alpha^2=\frac{1.9 \times 10^{-4}}{0.02}\)
or \(\alpha=\sqrt{95} \times 10^{-2}=9.75 \times 10^{-2}\).
The degree of dissociation of formic acid = 0.0097.
Acid Base Equilibrium
Acids and bases are also electrolytes. You are quite familiar with some properties of acids and bases. You know, for example, that adds turn blue litmus red and that bases (also called alkalis) turn red litmus blue.
You know that adds react with active metals like zinc and magnesium to liberate hydrogen. You know that acids are sour and corrosive and that bases are bitter and slippery. And you also know that adds and bases neutralise each other.
This is essentially the concept chemists had of acids and bases in the olden days. You could call this concept, based on certain properties of adds and bases, the dassical concept.
Arrhenius theory: In 1884, S A Arrhenius, a Swedish chemist, proposed a new definition of acids and bases. It was based on his general theory of ionisation of electrolytes, which basically said that an electrolyte, on dissolution in water, dissociated into positively and negatively charged ions.
Arrhenius defined an acid as a substance which produces hydrogen ions (H+) when mixed with water. Bases, according to Arrhenius, are substances which produce hydroxyl ions (OH–) when mixed with water. And the neutralisation of adds and bases is a reaction between H+ and OH– ions.
⇒ \(\mathrm{H}^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq}) \rightleftharpoons \mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
Strong acids like HCl, HNO3 and H2SO4 dissociate completely when dissolved in water.
⇒ \(\mathrm{HCl} \stackrel{\text { water }}{\longrightarrow} \mathrm{H}^{+}+\mathrm{Cl}^{-}\)
⇒ \(\mathrm{HNO}_3 \stackrel{\text { water }}{\longrightarrow} \mathrm{H}^{+}+\mathrm{NO}_3\)
Weak acids like acetic acid, carbonic acid and phosphoric acid dissociate only to a small extent when dissolved in water.
⇒ \(\mathrm{CH}_3 \mathrm{COOH} \stackrel{\text { water }}{\rightleftharpoons} \mathrm{CH}_3 \mathrm{COO}^{-}+\mathrm{H}^{+}\)
⇒ \(\mathrm{H}_2 \mathrm{CO}_3 \stackrel{\text { water }}{\rightleftharpoons} 2 \mathrm{H}^{+}+\mathrm{CO}_3^{2-} \)
⇒ \(\mathrm{H}_3 \mathrm{PO}_4 \stackrel{\text { water }}{\rightleftharpoons}{=} 3 \mathrm{H}^{+}+\mathrm{PO}_4^{3-}\)
Bases like NaOH and KOH are strong bases because they dissociate completely.
⇒ \(\mathrm{NaOH} \stackrel{\text { water }}{\longrightarrow} \mathrm{Na}^{+}+\mathrm{OH}^{-}\)
⇒ \(\mathrm{KOH} \stackrel{\text { water }}{\longrightarrow} \mathrm{K}^{+}+\mathrm{OH}^{-}\)
Substances like NH4OH, Ca(OH)2 and Al(OH)3 dissociate to a small extent and are called weak bases.
⇒ \(\mathrm{NH}_4 \mathrm{OH} \stackrel{\text { water }}{\rightleftharpoons} \mathrm{NH}_4^{+}+\mathrm{OH}^{-}\)
⇒ \(\mathrm{Ca}(\mathrm{OH})_2 \stackrel{\text { water }}{\rightleftharpoons} \mathrm{Ca}^{2+}+2 \mathrm{OH}^{-}\)
⇒ \(\mathrm{Al}(\mathrm{OH})_3 \stackrel{\text { water }}{\rightleftharpoons} \mathrm{Al}^{3+}+3 \mathrm{OH}^{-}\)
- According to Arrhenius, acids produce H+ ions and bases produce OH– ions when dissolved in water.
- Actually, the H+ ion, formed by the loss of an electron from the hydrogen atom, is only an unshielded proton.
- This proton cannot exist independently since its charge density is very high.
In an aqueous solution, it binds itself with one of the two lone pairs available on the water molecule to form a hydronium ion (H3O+).
⇒ \(\mathrm{H}^{+}+\mathrm{H}_2 \mathrm{O} \longrightarrow \mathrm{H}_3 \mathrm{O}^{+}\)
For the sake of convenience, the hydrated proton (hydronium ion) is represented as H+ (aq). In an aqueous solution, the H+ and OH– ions exist as hydrated ions.
Limitations of Arrhenius’s theory This concept of acids and bases could explain neutralisation, salt hydrolysis and the strength of adds and bases, but it had a few drawbacks.
- This theory cannot explain the basic nature of substances like NH3, Na2CO3 and CaO, which do not possess a hydroxyl (OH–) group. Nor cannot it explain the basic nature of CO2, SO2, SO3, etc., which do not contain hydrogen.
- Another limitation of this theory is its inability to explain reactions between acidic and basic substances in the absence of water. Two examples follow.
⇒ \(\mathrm{SO}_3(\mathrm{~g})+\mathrm{CaO}(\mathrm{s}) \longrightarrow \mathrm{CaSO}_4(\mathrm{~s})\)
⇒ \(\mathrm{NH}_3(\mathrm{~g})+\mathrm{HCl}(\mathrm{g}) \longrightarrow \mathrm{NH}_4 \mathrm{Cl}(\mathrm{s})\)
Bronsted-Lowry theory: In 1923, a Danish chemist, I N Bronsted, and a British chemist, T M Lowry, independently put forward a more general theory of acids and bases.
According to this theory, an acid is a proton donor and a base is a proton acceptor. This definition has some very interesting implications. To understand these, let us consider some examples.
⇒ \(\mathrm{HCl}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{Cl}^{-}\)
⇒ \(\mathrm{CH}_3 \mathrm{COOH}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{CH}_3 \mathrm{COO}^{-}\)
⇒ \(\mathrm{CO}_3^{2-}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{HCO}_3^{-}+\mathrm{OH}^{-}\)
⇒ \(\mathrm{NH}_3+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{NH}_4^{+}+\mathrm{OH}^{-}\)
⇒ \(\mathrm{NH}_4^{+}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{NH}_3\)
⇒ \(\mathrm{HCl}+\mathrm{NH}_3 \rightleftharpoons \mathrm{NH}_4^{+}+\mathrm{Cl}^{-}\)
Consider the first two reactions. HCl is an acid because it donates one proton to become Cl– and H2O is a base because it accepts a proton to become H3O+.
- Similarly, in the second equation, CH3COOH is an acid and H2O is a base. Now consider the next two reactions. In these, water acts as an acid.
- It donates a proton to become OH–. This means that the same substance can act as an acid or a base. Such substances which can act both as an acid and a base are called amphoteric.
- This definition of acids and bases does not restrict itself to neutral molecules. Even ions can act as acids or bases. For example, CO32- in the third equation acts as a base by accepting a proton.
Nor is it necessary for an acid-base reaction to take place in an aqueous medium. For example, in the sixth equation HCI acts as an acid by donating a proton and NH3 acts as a base by accepting a proton.
- The presence of the hydroxyl group (OH–) is not necessary for a substance to act as a base. Similarly, a substance can act as an acid even if it does not contain hydrogen.
- For example, NH3 acts as a base in the fourth reaction. All that is necessary for an acid-base reaction is that one substance should be able to donate a proton and the other should be able to accept the proton.
- A corollary is that a substance which acts as an acid in the presence of a proton-acceptor, will not act in the same way in the absence of a proton-acceptor.
- For example, though acetic acid acts as an acid in an aqueous solution (second equation), it does not do so in a benzene solution because benzene is not a proton-acceptor.
The Bronsted-Lowry concept of acids and bases has a very interesting consequence. When a substance acts as an acid it donates a proton. But on donating a proton, it becomes a base because it becomes capable of accepting a proton. Let us take the case of NH3 and H2O.
⇒ \(\mathrm{H}_2 \mathrm{O}+\mathrm{NH}_3 \rightleftharpoons \mathrm{NH}_4^{+}+\mathrm{OH}^{-}\)
Here water acts as an acid by donating a proton and becomes OH–. Now OH– can accept a proton, so it can act as a base.
- Similarly, NH3 acts as a base by accepting a proton and becomes NH+4, and NH+4 has a proton to donate, so it is an acid.
- Now do you see how every acid turns into a base by donating a proton, and every base turns into an add by accepting a proton.
- The base formed when an acid donates a proton is called the conjugate base of the add and the acid formed when a base accepts a proton is called the conjugate acid of the base.
In the reaction between NH3 and H2O, OH– is the conjugate base of H2O and NH4 is the conjugate acid of NH3.
Thus, there are two acid-base pairs in the reaction and these are called conjugate acid-base pairs. A few examples will make this clearer.
⇒ \(\text { Acid } 1+\text { base } 2 \rightleftharpoons \text { acid } 2+\text { base } 1\)
⇒ \(\mathrm{HCl}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{Cl}^{-}\)
⇒ \(\mathrm{H}_2 \mathrm{O}+\mathrm{CO}_3^{2-} \rightleftharpoons \mathrm{HCO}_3^{-}+\mathrm{OH}^{-}\)
⇒ \(\mathrm{HCO}_3^{-}+\mathrm{NH}_3 \rightleftharpoons \mathrm{NH}_4^{+}+\mathrm{CO}_3^{2-}\)
⇒ \(\mathrm{NH}_4^{+}+\mathrm{CH}_3 \mathrm{COO}^{-} \rightleftharpoons \mathrm{CH}_3 \mathrm{COOH}+\mathrm{NH}_3\)
- A strong add has a strong tendency to donate a proton but its conjugate base does not have an equally strong tendency to accept a proton.
- The conjugate base of a strong acid is weak and the conjugate base of a weak acid is strong. Also, if two acids are mixed, the weaker acid acts as a base.
⇒ \(\underset{\text { (perchloric acid) }}{\mathrm{HClO}_4}+\mathrm{H}_2 \mathrm{SO}_4 \rightleftharpoons \mathrm{H}_3 \mathrm{SO}_4^{+}+\mathrm{ClO}_4^{-}\)
In this reaction, H2SO4 acts as a base with respect to the stronger acid, HClO4. Thus, H2SO4 is amphoteric or amphiprotic. Certain acids can lose two or three protons. They are called diprotic (H2SO4) or triprotic (H3PO4) adds.
Strengths of acids and bases: The strength of an acid or base depends upon its readiness to lose or gain a proton. Experimentally, the strength of an acid or base is measured by its ionisation constant or dissociation constant.
- A strong acid like HCl, HNO3, H2So4, HBr, HI and HClO4 dissociates almost completely in water. This means the molar concentration of H3O+ ions in an aqueous solution of a strong acid is almost the same as that of the acid itself.
- But this is not the case for weak acids and we can compare the strengths of such acids if we know the dissociation constants at a particular temperature.
- The equilibrium constant for the dissociation of an acid is called its acid dissociation constant or acid ionisation constant and is denoted by Ka.
- Similarly, the base dissociation or base ionisation constant is Kb.
Consider a weak acid HA and suppose its dissociation equilibrium can be represented as follows.
⇒ \(\mathrm{HA}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{A}^{-}(\mathrm{aq})\)
At equilibrium, \(K_{\mathrm{a}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]\left[\mathrm{H}_2 \mathrm{O}\right]}\). Here [H2O] = 1 as it is in a very large quantity and remains almost unchanged.
Suppose the initial concentration of HA is C mol L-1 and its degree of dissociation is α. Then
⇒ \(K_{\mathrm{a}}=\frac{(C \alpha)^2}{C(1-\alpha)}=C \alpha^2\) (neglecting a in comparison with 1)
or \(\alpha =\sqrt{\frac{K_{\mathrm{a}}}{C}}\).
Thus, the degree of dissociation is inversely proportional to the square root of the concentration. Suppose we consider two acids of equimolar concentration (i.e., C is the same for both) and degrees of dissociation α1 and α2.
Then, \(\frac{\alpha_1}{\alpha_2}=\sqrt{\frac{K_{a_1}}{K_{a_2}}}\)
However, the degree of dissociation of an acid is a measure of its strength.
Therefore, \(\frac{\text { strength of acid }}{\text { strength of } \text { acid }_2}=\sqrt{\frac{K_{\mathrm{a}_1}}{K_{\mathrm{a}_2}}} \text {. }\)
- So, we may say that at a given temperature, the ionisation or dissociation constant of an add is the measure of
its strength.
- As you already know, the equilibrium constant is a dimensionless quantity so Ka is also without any unit.
- Concentrations of all the speeds at ionisation equilibrium are measured with respect to the standard state concentration of 1 M.
- The larger the value of Ka, the stronger is the acid. Weak acids, however, show a great variation in their strengths. For instance, both formic add and hydrocyanic add are weak but their ionisation constants are very different.
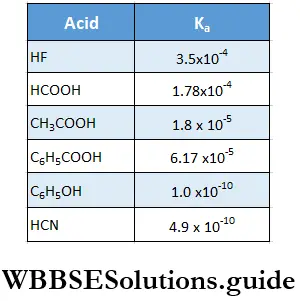
Example: The ionisation constants of formic acid (HCOOH) and hydrocyanic acid (HCN) are 1.77 x 1024 and 4.9 x 10-10 respectively. Compare their strengths.
Solution:
Given
The ionisation constants of formic acid (HCOOH) and hydrocyanic acid (HCN) are 1.77 x 1024 and 4.9 x 10-10 respectively.
\(\frac{\text { Strength of formic acid }}{\text { Strength of hydrocyanic acid }}=\sqrt{\frac{K_{\mathrm{a}}(\mathrm{HCOOH})}{K_{\mathrm{a}}(\mathrm{HCN})}}=\sqrt{\frac{1.77 \times 10^{-4}}{4.9 \times 10^{-10}}}=601 \text {. }\)
Therefore, formic acid is 601 times stronger than hydrocyanic acid.
The relative strengths of bases can be determined in a similar manner. First, let us consider the equilibria involved in solutions of weak bases. If B is the base, the equilibrium may be represented as
⇒ \(\mathrm{B}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{BH}^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)
The equilibrium constant is Kb and is called base dissociation constant. The higher the value of Kb, the stronger is the base.
“Dynamic equilibrium in reversible reactions with explanation”
⇒ \(K_{\mathrm{b}}=\frac{\left[\mathrm{BH}^{+}\right]\left[\mathrm{OH}^{-}\right]}{[\mathrm{B}]}\)
If C is the molar concentration of a base and α is its degree of dissociation \(\alpha=\sqrt{\frac{K_b}{C}},\)
where Kb is the dissociation constant of the base.
If we take two bases b1 and b2 of equimolar concentrations and dissociation constants \(K_{b_1} \text { and } K_{b_2}\) then
∴ \(\frac{\text { strength of base }}{\text { strength of base }{ }_2}=\sqrt{\frac{K_{b_1}}{K_{b_2}}} \text {. }\)
Relation between Ka and Kb We have seen that the strength of an acid (or a base) is expressed by its Ka (or Kb). For a conjugate acid-base pair, the two equilibrium constants are related in a simple manner.
Let us consider the case of the acid-base pair NH4 and NH3. The equilibrium for both acid and base may be represented as
⇒ \(\mathrm{NH}_4^{+}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{NH}_3(\mathrm{aq})\)
∴ \(K_{\mathrm{a}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{NH}_3\right]}{\left[\mathrm{NH}_4^{+}\right]}=5.6 \times 10^{-10}\)
∴ \(\mathrm{NH}_3(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{NH}_4^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)
∴ \(K_{\mathrm{b}}=\frac{\left[\mathrm{NH}_4^{+}\right]\left[\mathrm{OH}^{-}\right]}{\left[\mathrm{NH}_3\right]}=1.8 \times 10^{-5}\).
On adding the two, we get the net reaction as \(2 \mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)
The sum of the two reactions is simply the dissociation of water, and its equilibrium constant,
⇒ \(K_{\mathrm{w}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{OH}^{-}\right]}{\left[\mathrm{H}_2 \mathrm{O}\right]^2}\)
or \(K_{\mathrm{w}}=\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{OH}^{-}\right]=1.0 \times 10^{-14}\).
The equilibrium constant for the net reaction is equal to the product of the equilibrium constants for the individual reactions.
⇒ \(K_{\mathrm{a}} \times K_{\mathrm{b}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{NH}_3\right]}{\left[\mathrm{NH}_4^{+}\right]} \times \frac{\left[\mathrm{NH}_4^{+}\right]\left[\mathrm{OH}^{-}\right]}{\left[\mathrm{NH}_3\right]}=\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{OH}^{-}\right]=K_w,\)
i. e., (5.6 x 10-10) x (18 x 10-5) = 10 x 10-14. The ionic product of water (Kw) is discussed later in the chapter.
Whenever an equation can be written as a sum of two or more equations, the equilibrium constant for the net reaction is the product of the equilibrium constants of all the individual reactions.
Di- and polybasic acids and di- and polyacidic bases Polybasic or polyprotic acids are those which have more than one ionisable proton. For example, sulphuric acid has two and phosphoric acid has three ionisable protons. The ionisation reactions for a dibasic acid like H2SO4 can written as
⇒ \(\mathrm{H}_2 \mathrm{SO}_4(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l})\rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{HSO}_4^{-}(\mathrm{aq}) \)
⇒ \(\mathrm{HSO}_4^{-}(\mathrm{aq}) \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{SO}_4^{2-}(\mathrm{aq})\)
The corresponding equilibrium constants for these reactions will be as follows.
⇒ \(K_{\mathrm{a}_1}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{HSO}_4^{-}\right]}{\left[\mathrm{H}_2 \mathrm{SO}_4\right]} \text { and } K_{\mathrm{a}_2}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{SO}_4^{2-}\right]}{\left[\mathrm{HSO}_4^{-}\right]} \text {. }\)
- As you can see, the dibasic acid H2SO4 has two ionisation constants. Similarly, tribasic acids like phosphoric acid will have three ionisation constants.
- Like polybasic acids, polyacidic bases also have the ionisation constants corresponding to the number of ionisation steps.
- For example, a diacidic base like ethylenediamine has two ionisation constants \(K_{b_1} \text { and } K_{b_2}\). Table gives some values of the ionisation constants of polyprotic acids.
- As you can seen in Table among all the ionisations in an individual polybasic acid the first ionisation constant is the largest followed by the second and then the third.
- This is because after the first ionisation, we are left with a negatively charged ion and it is difficult to remove a proton from a negative ion.
Factors affecting acid strength: We have already seen how to compare the strengths of two acids. But why is one acid stronger than the other? Put simply, the acid strength is determined by the strength and polarity of the H—A bond.
- The strength of the bond depends on the enthalpy change associated with the dissociation of the HA molecule into H and A.
- The polarity of the H—A bond depends on the electronegativity of A and the ease with which electron transfer can occur from H to A resulting in H+ and A– ions. The weaker and more polar the HA bond, the stronger is the acid.
- Let us consider the acids of the halogen series HF, HCl, HBr and HI.
- The variation in polarity is not very significant while we move from HF to Til but there is a considerable difference in the bond strength, which is the deciding factor here.
You already know that atomic size increases on moving down a group. Thus, the size increases from F to I and so the bond strength decreases, increasing the acidity from HF to HI.
- This trend is generally true of acids in the same group of the periodic table. When we go down the group from O to Te, the acid strength increases as H2O<H2S<H2Se<H2Te.
- For acids in the same row of the periodic table, the variation in bond strength is insignificant and the polarity of the bond is the deciding factor for acid strengths.
- Let us consider the hydrides of a few elements in the second period—CH4, NH3, H2O and HF. As we move from C to F, electronegativity increases and hence the added strength also increases from CH4 to HF.
- The electronegativities given are C, N, O and F respectively.
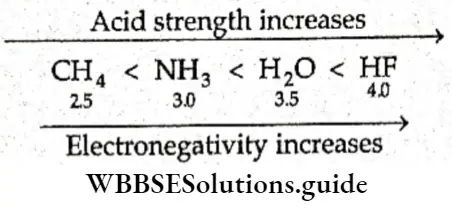
Oxoacids Oxoacids such as H2SO4, H2CO3 and HNO3 have an O—H bond and the acid strength is dependent on the strength of this bond. Any factor that weakens the O—H bond or increases its polarity, increases the strength of the acid.
- In these acids, the nonmetallic atom is bonded on one side to the oxygen of O—H. The electronegativity of this atom and its oxidation number influences the acid strength.
- In the case of oxoacids containing the same number of O—H groups and the same number of O atoms, as the electronegativity of the nonmetallic atom increases, the acid strength increases.
- For example, consider HOCl, HOBr and HOI. The electronegativity decreases from Cl to I, and hence add strength increases from HOI to HOBr to HOCl.
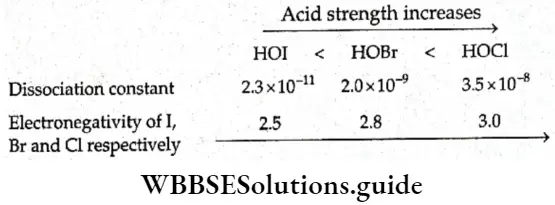
With the increase in the electronegativity of the nonmetallic atom attached to the O—H bond, the bonded electron cloud is pulled more towards the atom, thus weakening the O—H bond, which then tends to break easily.
If the oxoacids contain the same nonmetallic atom but different numbers of oxygen atoms, then as the oxidation number of the atom increases, the acid strength increases. For example, among the oxoacids of chlorine, the strength of the acids increases as
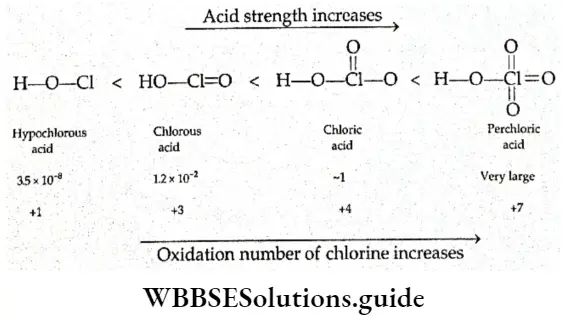
Common-Ion effect in the ionisation of acids and bases: You are already aware how acetic acid ionises in water. You also know how to calculate the degree of dissociation of this acid. The equilibriun involved is \(\mathrm{CH}_3 \mathrm{COOH}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{CH}_3 \mathrm{COO}^{-}+\mathrm{H}_3 \mathrm{O}^{+}\)
∴ \(K_{\mathrm{a}}=\frac{\left[\mathrm{CH}_3 \mathrm{COO}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{\left[\mathrm{CH}_3 \mathrm{COOH}\right]} .\)
If we add sodium acetate (the salt of the conjugate base of acetic acid) to the acetic acid solution, the hydrogen ion concentration changes (it decreases).
- This is because the addition of acetate ions causes an increase in the concentration of the products and the equilibrium shifts to the left-hand side, reducing the dissociation of acetic acid.
- This is according to Le Chatelier’s principle. This is called the common ion effect, which is simply the shift in equilibrium on the addition of a substance that provides more of an ion already involved in the equilibrium. Therefore, the concentration of H3O+ decreases.
Suppose acetate ions of concentration 0.1 M are added a 0.1-M acetic acid solution. Then the initial concentrations of the acetate ion and acetic acid solution are both 0.1 M.
Let the concentration of the acetic acid dissociated be x. Then the concentrations of the species present can be given as follows:
⇒ \(\mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})+\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})\)
Initial conc. \( 0.1 0.1 \sim 0\)
Change \(-x +x +x\)
Equilibrium conc. \(0.1-x 0.1+x x\)
⇒ \(K_{\mathrm{a}}=18 \times 10^{-5}=\frac{\left[\mathrm{CH}_3 \mathrm{COO}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{\left[\mathrm{CH}_3 \mathrm{COOH}\right]}\)
= \(\frac{(0.1+x)(x)}{0.1-x}\).
Since Ka is small for a very weak acid, x is small compared to 0.1 and we can make the approximation that 0.1 +x ≈ 0.1 ≈ 0.1-x.
∴ \(K_{\mathrm{a}}=18 \times 10^{-5}=\frac{x(0.1)}{0.1}=x\)
or x = [H3O+] = 1.8 x 10-5 M.
Example: Calculate the hydronium iott concentration of a 0.1-M solution of acetic acid, given Ka = 1.8 x 10-5.
Solution:
⇒ \(\mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})+\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})\)
⇒ \(K_{\mathrm{a}}=\frac{\left[\mathrm{CH}_3 \mathrm{COO}^{-}\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\right.}{\left[\mathrm{CH}_3 \mathrm{COOH}\right]}\).
If x is the concentration of CH3COOH that dissociates, then the concentrations for the various species in the solution are as follows.
⇒ \(\mathrm{CH}_3 \mathrm{COOH}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{CH}_3 \mathrm{COO}^{-}+\mathrm{H}_3 \mathrm{O}^{+}\)
Initial conc. \(0.1 0 0\)
Change \(-x +x +x\)
Equilibrium conc. \(0.1-x x x\)
Substituting the values at equilibrium in the expression for Ka,
⇒ \(K_{\mathrm{a}}=\frac{x \times x}{0.1-x}=\frac{x^2}{0.1-x}\)
This will result in a quadratic equation in x. However, it may be simplified by assuming that x << 0.1
Hence, \(K_{\mathrm{a}}=\frac{x^2}{0.1}\)
⇒ x2 = 0.1 x Ka = 0.1 x 1.8 x 10-5
x = 1.34 x 10-3
∴ [H3O+] = 1.34 x 10-3
Limitations of Bronsted-Lowry theory The Bronsted-Lowry theory could explain many reactions which the Arrhenius theory could not. However, it still could not account for certain acid-base reactions which do not involve the transfer of a proton.
1. Reactions between acidic oxides like CO2, SO2 and SO3 and basic oxides like CaO, BaO and MgO cannot be explained by the theory of proton transfer. Such reactions take place in the absence of a solvent.
⇒ \(\mathrm{CaO}+\mathrm{SO}_3 \longrightarrow \mathrm{CaSO}_4\)
⇒ \(\mathrm{MgO}+\mathrm{CO}_2 \longrightarrow \mathrm{MgCO}_3\)
2. The acidic behaviour of compounds like BF3 and AlCl3 cannot be explained by the Bronsted-Lowry theory.
Lewis theory: In 1923, G N Lewis, an American chemist, extended the concept of adds and bases further.
- According to the Lewis theory, an acid is a substance which can accept a pair of electrons and a base is a substance which can donate a pair of electrons.
- While this definition encompasses most add-base reactions, it has the drawback of being so general as to indude other types of reactions in its ambit.
- According to the Lewis theory, an acid-base reaction involves the donation of a pair of electrons by a base to an add, leading to the formation of a coordinate bond between them.
Lewis bases Lewis bases can be of two types,
- Neutral molecules like H2O, RNH4, NH3 and ROH, in which
one atom has at least one unshared pair of electrons, can act as Lewis bases,
- All negative ions (F–, Cl–, OH–, CN–, etc.) can behave as Lewis bases.
Lewis acids Species with vacant orbitals in the valence shell of one of the atoms can act as Lewis acids. Some examples of Lewis acids are as follows.
1. Simple cations like Ag+, Cu2+ and Fe3+ can accept a pair of electrons and act as Lewis acids.
2. Molecules which have an atom with an incomplete octet can behave as Lewis acids, for example, BF3 and AlCl3.
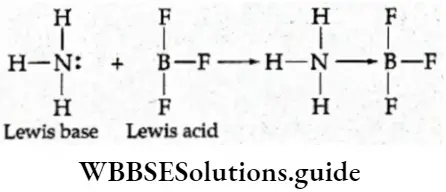
3. Molecules in which the central atom has vacant orbitals may acquire more than an octet of valence electrons, for example, SnCl4, SiF4 and PF5, and can act as Lewis acids.
⇒ \(\underset{\substack{\text { Lewis acid } \\ \text { acid }}}{\mathrm{SiF}_4}+\underset{\text { Lewis base }}{2 \mathrm{~F}^{-}} \longrightarrow \mathrm{SiF}_6^{2-}\)
4. Molecules which have a multiple bond between two atoms of different electronegativities, for example, CO2 and SO2, also behave as Lewis acids.
In the CO2 (O=C=O) molecule, carbon being less electronegative than oxygen, acquires a slight positive charge (because one n electron pair shifts more towards oxygen) and can, thus, accept a pair of electrons.
⇒ \(\underset{\substack{\text { Lewis } \\ \text { acid }}}{\mathrm{CO}_2}+\underset{\substack{\text { Lewis } \\ \text { base }}}{\mathrm{OH}^{-}} \longrightarrow \mathrm{HCO}_3^{-}\)
- While it is true that the definition of acids and bases proposed by Lewis is more general than the other definitions, it does not mean that it encompasses all acid-base reactions.
- There are substances, for instance, which are Bronsted acids but not Leivis acids. Bronsted acids like HCl and H2SO4 can donate a proton but are not capable of accepting a pair of electrons, so they are not Lewis acids.
However, all Bronsted bases are also Lewis bases because a species capable of donating an electron pair (Lewis base) also has the tendency to accept a proton (Bronsted base).
⇒ \(\mathrm{NH}_3+\mathrm{H}_2 \mathrm{O} \longrightarrow \mathrm{NH}_4^{+}+\mathrm{OH}^{-}\)
Here NH3 is a Lewis base as well as n Bronsted base but H2O is a Bronsted acid, but not a Lewis add.
Limitations of Lewis theory The Lewis theory of adds and bases can explain the acidic and basic nature of many substances which cannot be explained by either the Bronsted-Lowry theory or the Arrhenius theory. Still it is not a flawless theory.
- This concept of adds and bases is so general that it labels all reactions leading to the formation of coordinate bonds as acid-base reactions.
- It does not explain the addic behaviour of adds like HCl and H2SO3 which do not form coordinate bonds with bases.
- The formation of coordination compounds is usually a slow process, so add-base reactions should be slow- according to the Lewis theory. But acid-base reactions are generally fast.
- The Lewis theory cannot be used to ascertain the relative strengths of adds and bases.
- The catalytic property of many adds is due to the H+(aq) ion. Since a Lewis add need not possess hydrogen, a Lewis acid need not have this property.
While dealing with acids and bases such as HCl, CH3COOH, NaOH and NH4OH in aqueous solutions, the Bronsted-LowTy concept is the most suitable and hence widely accepted.
Self Ionisation Of Water
So far we have considered water as a solvent and seen that it has both acidic and basic properties, i.e., it is amphoteric in nature. We will be using the Bronsted-Low’ry concept to understand the ionisation of water. In the presence of an add, it acts as a base while in the presence of a base, it acts as an acid.
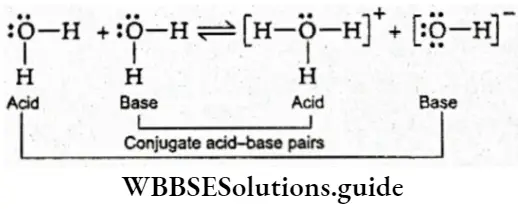
In pure water, one molecule of water can donate a proton to another molecule of water in a reaction in which water acts as acid and base simultaneously. There are always some H3O+ and OH– ions present in pure water.
or \(\mathrm{H}_2 \mathrm{O}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{OH}^{-}\)
This is referred to as dissociation self-ionisation or auto-ionisation of water and is characterised by the equilibrium constant,
or, \(K_{\mathrm{c}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{OH}^{-}\right]}{\left[\mathrm{H}_2 \mathrm{O}\right]\left[\mathrm{H}_2 \mathrm{O}\right]}\)
Multiplying the denominator of the above expression ([H2O]2) by Kc (as H2O is in excess and is essentially constant), we get K=[H2O+][OH–]
Kw is called the ionic product of water. It is a constant at a particular temperature. At 298 K its value is 1.0 x 1014 mol2 L-2. Such a small value of Kw indicates that the auto-ionisation of water does not occur to a large extent.
In fact, the reverse reaction, the formation of water by the reaction of H3O+ and OH– essentially goes to completion as its equilibrium constant is
⇒ \(\frac{1}{1 \times 10^{-14}}=1 \times 10^{14}\)(As stated earlier the units have been dropped.)
Kw increases with temperature because the degree of ionisation of water increases with temperature.
In pure water both H3O+ and [OH–] are equal.
⇒ \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\left[\mathrm{OH}^{-}\right]=\sqrt{1 \times 10^{-14}}=1 \times 10^{-7} \text { at } 298 \mathrm{~K} \text {. }\)
We can calculate the extent of dissociation of water molecules by finding the concentration of dissociated and undissociated water.
The concentration of dissociated water is simply the concentration of [H3O+] or [OH–]
Therefore concentration of dissociated water =1 x 10-7 M at 298 K.
The molar concentration of pure water can be calculated from its density and volume.
Now, number of moles = \(\frac{\text { mass }}{\text { molar mass }}=\frac{D \times V}{\text { molar mass }}\) (since mass = density x volume)
Concentration of undissociated water =\(\left(\frac{1000 \mathrm{~g}}{\mathrm{~L}}\right)\left(\frac{1 \mathrm{~mol}}{18 \mathrm{~g}}\right)=55.4 \mathrm{~mol} \mathrm{~L}^{-1}\)
(The density of pure water is 1000 gL-1 and its molar mass is 18.0 g mol-1.)
Therefore degree of dissociation = \(\frac{\text { conc. of dissociated water }}{\text { conc. of undissociated water }}\)
= \(\frac{1 \times 10^{-7}}{55.4}=18 \times 10^{-9} \approx 2 \times 10^{-9} \text { or } \frac{2}{10^9} \text {. }\)
This means that out of 109 molecules of water, two molecules are ionised.
Aqueous solutions of acids and bases: The concentrations of H3O+ ions and OH– ions are the same in pure water. When some acid or base is added to water, these concentrations do not remain equal, but the value of Kw remains the same (at a particular temperature).
- If some acid is added to water, the concentration of H3O+ ions will increase.
- Then according to Le Chatelier’s principle, the system will try to counteract the change and the reverse reaction will be favoured, i.e., H3O+ ions will combine with OH– ions to form water molecules.
- In other words, the equilibrium will shift backwards, so that the value of Kw may remain the same.
At the new equilibrium, the concentration of OH– ions will be less than the concentration of H3O+ ions. The concentration of OH– ions can be obtained from the following relation.
⇒ \(\left[\mathrm{OH}^{-}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}\)
Similarly, the addition of a base will increase the concentration of OH– ions and decrease the concentration of H3O+ ions.
The concentration of H3O+ ions in an aqueous solution of a base can be obtained from the following expression.
⇒ \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{OH}^{-}\right]} .\)
In pure water or neutral solutions [H3O+] = [OH–].
In acidic solutions [H3O+]> [OH–].
In basic solutions [H3O+]<[OH–].
According to Arrhenius, a strong acid or a strong base completely dissociates into hydrogen ions and the corresponding anions or hydroxyl ions and the corresponding cations (as the case may be).
Thus, a 0.1-M HCl solution will dissociate to give 0.1-M H+ ions and 0.1-M Cl– ions.
Example: Calculate the hydronium ion and hydroxyl ion concentrations in
- a 0.001-M HCl solution,
- a 0.01-M HNO3 solution,
- a 0.01-M NaOH solution,
- a 0.01-M Ba(OH)2 solution,
- a 0.1-M HCOOH which is 10% ionised and
- a solution containing 3.65 x 10-3 g of HCl per 100 mL.
Solution:
1. HCl is a strong acid which ionises completely in water, so[H3O+] = 0.00 1 M = 1 x 10-3 mol L-1.
The equilibrium can be represented as follows.
⇒ \(\mathrm{HCl}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{Cl}^{-}\)
⇒ \(K_{\mathrm{w}}=\left[\mathrm{OH}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\)
⇒ \({\left[\mathrm{OH}^{-}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}=\frac{1 \times 10^{-14}}{10^{-3}}=1 \times 10^{-11} \mathrm{~mol} \mathrm{~L}^{-1}}\)
2. The equilibrium in this case can be represented as \(\mathrm{HNO}_3+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{NO}_3^{-}\)
Being a strong acid HN03 too dissociates completely and
⇒ \({\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\left[\mathrm{HNO}_3\right]=0.01 \mathrm{M}=1 \times 10^{-2} \mathrm{~mol} \mathrm{~L}^{-1} . }\)
∴ \(\quad{\left[\mathrm{OH}^{-}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{H}_0 \mathrm{O}^{+}\right.}=\frac{1 \times 10^{-14}}{10^{-2}}=1 \times 10^{-12} \mathrm{~mol} \mathrm{~L}^{-1}.}\)
3. Since NaOH is a strong base it ionises completely in water.
∴ \(\quad\left[\mathrm{OH}^{-}\right]=[\mathrm{NaOH}]=0.01 \mathrm{M}=10^{-2} \mathrm{~mol} \mathrm{~L}^{-1}\)
∴ \(\quad\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{OH}^{-}\right]}=\frac{1 \times 10^{-14}}{10^{-2}}=1 \times 10^{-12} \mathrm{~mol} \mathrm{~L}^{-1}\).
4. Since Ba(OH)2 is completely ionised and its acidity is 2, i.e.,1 mol of Ba(OH)2 gives 2 mol of OH– ions,
⇒ \(\mathrm{Ba}(\mathrm{OH})_2 \longrightarrow \mathrm{Ba}^{2+}+2 \mathrm{OH}^{-}\)
⇒ \({\left[\mathrm{OH}^{-}\right]=2\left[\mathrm{Ba}(\mathrm{OH})_2\right]=2 \times 0.01=0.02 \mathrm{~mol} \mathrm{~L}^{-1} .}\)
∴ \({\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{OH}^{-}\right]}=\frac{1 \times 10^{-14}}{2 \times 10^{-2}}=5 \times 10^{-13} \mathrm{~mol} \mathrm{~L}^{-1} .}\)
5. HCOOH is a weak acid whose degree of dissociation (α) is 10% (given).
⇒ \(\underset{\mathrm{C}(1-\alpha)}{\mathrm{HCOOH}}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \underset{\mathrm{C} \alpha}{\mathrm{H}_3 \mathrm{O}^{+}}+\underset{\mathrm{C} \alpha}{\mathrm{HCOO}^{-}}\)
⇒ \({\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\mathrm{C} \alpha=0.1 \times 0.1=0.01=10^{-2} \mathrm{~mol} \mathrm{~L}^{-1} .}\)
⇒ \({\left[\mathrm{OH}^{-}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}=\frac{1 \times 10^{-14}}{10^{-2}}=10^{-12} \mathrm{~mol} \mathrm{~L}^{-1} .}\)
6. Molarity of a solution = no. of moles in1 L of solution.
The given solution contains 3.65 x 10-3 g of HCl in 100 mL or 0.1 L.
∴ M = \(\frac{3.65 \times 10^{-3}}{36.5 \times 0.1}\) [mol. wt. of HCl = 36.5] = 1 x 10-3
HCl is a strong acid.
∴ \({\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=[\mathrm{HCl}]=1 \times 10^{-3} \mathrm{~mol} \mathrm{~L}^{-1} .}\)
∴ \({\left[\mathrm{OH}^{-}\right]=\frac{1 \times 10^{-14}}{10^{-3}}=1 \times 10^{-11} \mathrm{~mol} \mathrm{~L}^{-1} .}\)
The pH scale: From our discussions on the ionisation of water so far, it must have become clear that Kw (ionic product) remains constant and that for a neutral solution [H3O+] is equal to [OH–], for an acidic solution [H3O+] > [OH–] and for a basic solution [H3O+]< [OH–].
- Surely then if we know the H3O+ ion concentration in a solution we would have a fair idea about its relative acidic or basic character.
- The hydrogen ion concentration may vary from, say, 1 M in a strong acid to 10-14 M in a strongly basic solution. Expressing H3O+ ion concentrations in this fashion would be rather cumbersome.
- In 1909, P L Sorensen, a Danish chemist, proposed a more convenient way of expressing the
- H3O+
- ion concentration in a solution. The scale proposed by Sorensen is called the pH (power of hydrogen) scale, which may be defined as
- The magnitude of the negative power to which 10 must be raised to express the hydronium ion concentration; or
- The negative logarithm (base 10) of the hydronium ion concentration in mol L-1.
- Say the hydronium ion concentration in a solution is 10-x Then its pH would be x, according to the first definition. This is simple enough and the first definition of pH seems to be apt.
- But what if the H3O+ ion concentration in some solution is 1.5 x 10-4 M? The first definition would not be so convenient.
- This is why the second definition is preferred. Actually, the second definition can be derived from the first.
Suppose [H3O+] = 10-x
Then log[H3O+] = log 10-x = -x log 10 = -x.
∴ x = pH = -log[H3O+].
If you happen to know the pH of a solution, you could easily calculate the concentration of H3O+ ions in it. [H3O+]= antilog (-pH).
The hydroxyl ion concentration in a solution can be expressed in terms of pOH.
pOH = – log[OH–].
For any solution at 298 K
Kw = [H3O+][OH–] = 10-14
∴ log[H3O+] + log[OH–] = logKw = log 10-14 =-14
or -log[H3O+]-log[OH–] = -logKw =14
or pH + pOH = pKw =14.
For a neutral solution, [H3O+] = [OH–] = 10-7.
∴ pH = -log 10-7 =7.
This brings us to something very useful.
For an acidic solution, pH < 7.
For a basic solution, pH > 7.
For a neutral solution, pH = 7.
For practical purposes, the pH range is taken as 0 to 14, though theoretically, it may be possible to have H3O+ ion concentrations of more than 10° M (i.e., 1 M) or less than 10-14 M.
The table gives the pH values of some common substances in everyday life.
Example 1. Calculate the pH value of a solution of
- 0.01-M HCl and
- 0.01-M NaOH.
Solution:
1. \(\mathrm{HCl}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \longrightarrow \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
Since HCl is completely ionised,
[H3O+] = [HCl] = 0.01 M = 1 x 10-2 mol L-1.
pH = – log[H3O+] = – log 1 x 10-2 = – [-2] = 2.
2. \(\mathrm{NaOH}(\mathrm{aq}) \longrightarrow \mathrm{Na}^{+}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)
NaOH is a strong base which ionises completely.
[OH–] = [NaOH] = 0.01 M = 10-2 mol L-1.
∴ \(K_{\mathrm{w}}=\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{OH}^{-}\right]\)
∴ \(\quad {\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{OH}^{-}\right]}=\frac{1 \times 10^{-14}}{1 \times 10^{-2}}=10^{-12} \mathrm{~mol} \mathrm{~L}^{-1} . }\)
∴ \(\quad \mathrm{pH}=-\log \left[\mathrm{H}_3 \mathrm{O}^{+}\right]=-\log 10^{-12}=-[-12]=12\).
Example 2. 1000 mL of a solution contains 6.3 g of nitric acid. What is the pH of the solution if the acid is completely dissociated?
Solution:
Given
1000 mL of a solution contains 6.3 g of nitric acid.
The concentration of HNO3 = 6.3 g L-1.
Molecular weight of HNO3 = 63.
∴ M = 6.3/6.3 = 0.1.
⇒ \(\mathrm{HNO}_3+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{NO}_3^{-}\)
Since the acid is completely dissociated
[H3O+] = [HNO3]=0.1M = 10-1 M.
pH = – log[H3O+] = – log 10-1 = 1.
- The pH scale is very useful in getting an idea about the relative acidic (or basic) strengths of solutions. The higher the pH, the lower the concentration of hydronium ions in the solution.
- If the pH of a solution changes by one unit, there is a tenfold change in the molar concentration of that solution. Here it is important for you to know that pH is a measure of ‘hydrogen ion activity’.
- At the present level of learning, for the sake of simplicity, we have been using hydrogen ion concentration as a measure of pH since we are considering ideal (dilute) behavior of solutions.
- However, many solutions deviate from ideal behaviour with rise in concentration. To overcome this problem, we use the concept of activity.
- Activity is a physical quantity, a thermodynamic function that can be used in place of concentration. Now consider a concentrated solution, for example, 1 M HCl.
Let us use both its activity and molar concentration to determine its pH.
⇒ \(a_{\mathrm{H}_3 \mathrm{O}^{+}} \text {(activity) }=\gamma\left[\mathrm{H}_3 \mathrm{O}^{+} \right.\)., where y is the activity coefficient, a dimensionless quantity.
The activity being 0.81 the pH value of the solution turns out to be 0.092. On the other hand, if we use the concentration, pH = – log[1] = 0.
The ‘p’ in pH indicates (-log) so that px means -log(x). This is very useful in calculations involving exponential numbers so that it has been extended to other species. The most widely used are pKw, pKa, and pKb.
PKa = -logKa; pKb =-logKb.
As you already know, for a conjugate acid-base pair, Ka x Kb = Kw.
Taking negative logarithm on both sides, (- log Ka) + (- log Kb ) = (- log Kw)
or PKa+pKb =pKw.
Measuring pH The pH of a solution can be measured with the help of pH papers, indicators, or pH meters. While pH papers and indicators give an approximate pH value of the solution in question, pH meters give the exact value.
- An acid-base indicator is a substance that changes colour in a specific pH range of about 2 pH units. To determine the pH of a solution, a few drops of the indicator are added to the solution.
- To make the determination easier, nowadays a universal indicator is available, which is simply a mixture of several indicators, to make approximate measurements in the pH range 3-10.
- The color change on the addition of the indicator is noted and compared with a color chart to know the pH of the given solution.
- Nowadays, pH meters are available for commercial purposes. They measure pH with high precision.
A typical pH meter consists of a special measuring probe (a glass electrode) connected to an electronic meter that measures and displays the pH reading.
Hydrolysis Of Salts
The term hydrolysis refers to any reaction in which water is one of the reactants. Water, being an amphiprotic solvent, can act both as an acid or a base. When a salt is dissolved in water, it dissociates into its ions. If the salt is represented as MA, then we have
⇒ \(\mathrm{MA}(\mathrm{aq}) \longrightarrow \mathrm{M}^{+}(\mathrm{aq})+\mathrm{A}^{-}(\mathrm{aq})\)
Now, water may interact with the ions in two ways:
⇒ \(\underset{\text { acid }}{\mathrm{M}^{+}(\mathrm{aq})}+\underset{\text { base }}{2 \mathrm{H}_2 \mathrm{O}(\mathrm{l})} \underset{\text { base }}{\mathrm{MOH}(\mathrm{aq})}+\underset{\text { adid }}{\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})}\) ….(1)
⇒ \(\underset{\text { base }}{\mathrm{A}^{-}(\mathrm{aq})}+\underset{\text { acid }}{\mathrm{H}_2 \mathrm{O}(\mathrm{l})} \underset{\text { add }}{\mathrm{HA}(\mathrm{aq})}+\underset{\text { base }}{\mathrm{OH}^{-}(\mathrm{aq})}\)…..(2)
As you can see in reaction (1), the base from which the cation is obtained is generated while in reaction (2), the acid from which the anion is obtained is generated. The salt MA is obtained by a neutralisation reaction between the base, MOH, and the acid, HA.
- A closer look at the reverse reactions shows that M+ is the conjugate acid of the base MOH while A– is the conjugate base of the acid HA.
- On dissolving a particular salt in water, whether reaction (1) or (2) or both will occur and to what extent is dependent upon the strengths of the acid and the base from which the salt has been formed.
- The strength of an acid is inversely proportional to that of its conjugate form. If HA is a strong acid, its conjugate base, A–, will be weak and hence reaction (2) will occur only insignificantly.
- Similarly, if the base MOH is strong, its conjugate acid M+ will be weak and reaction (1) will occur to a very small extent.
- If both M+ and A– are obtained from a strong base and a strong acid respectively, hydrolysis occurs to a negligible extent or we may say that it does not occur at all. One such example is NaCl.
- Na+ is obtained from NaOH, a strong base, and Cl– is obtained from HCl, a strong acid.
- A solution of NaCl will not show any hydrolysis and the species present in solution will be only Na+ (aq), Cl–(aq), and H2O. Hence the pH will be 7 and the solution is neutral.
When a salt dissociates into ions on dissolution in water, the ion which is the conjugate of a strong acid/base does not hydrolyse.
“Chemical equilibrium constant and its significance”
For example, sodium acetate, CH3COONa (formed from a strong base and a weak acid), will dissociate in water as \(\mathrm{CH}_3 \mathrm{COONa}(\mathrm{aq}) \longrightarrow \mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})+\mathrm{Na}^{+}(\mathrm{aq}).\)
Here CH3COO– is the conjugate base of the weak acid CH3COOH. Therefore it will hydrolyse forming OH ions in the solution.
⇒ \(\mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{OH}^{-}(\mathrm{aq})\)….(3)
However, Na+ will not hydrolyse as it is obtained from a strong base, NaOH. Therefore, the solution will be basic due to the hydrolysis reaction (3), in which hydroxyl ions are produced.
Similarly, NH4Cl (formed from a strong acid and a weak base) dissociates as \(\mathrm{NH}_4 \mathrm{Cl}(\mathrm{aq}) \rightleftharpoons \mathrm{NH}_4^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
In this case NH4+ is the conjugate acid of the weak base NH3 and hence undergoes hydrolysis as \(\underset{\text { acid }}{\mathrm{NH}_4^{+}(\mathrm{aq})}+\underset{\text { base }}{2 \mathrm{H}_2 \mathrm{O}(\mathrm{l})} \rightleftharpoons \underset{\text { base }}{\mathrm{NH}_4 \mathrm{OH}(\mathrm{aq})}+\underset{\text { acid }}{\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})}\)…(4)
Cl– will not undergo hydrolysis as it is obtained from HCl (strong acid). As a result, a solution of NH4Cl will be acidic due to reaction (4), where H3O+ is produced.
- Now let us see what happens when ammonium acetate, CH3COONH4, is dissolved in water.
- Since the salt is formed from the weak acid CH3COOH and the weak base NH4OH, both the ions (CH3COO– and NH4) formed will hydrolyse.
- Since both OH– and H3O+ are produced during hydrolysis, the pH of the solution will depend on the comparative strengths of the parent acid (Ka) and base (Kb) (from which the salt is formed).
- If Ka < Kb, the solution will be alkaline as then the conjugate base of the acid (A–) is stronger and will hydrolyse more as compared to the conjugate acid of the base (M+).
If Ka >Kb, the solution will be acidic. However, if Ka = Kb, the solution will be neutral. It is interesting to note that the pH of such solutions is independent of the concentration of the salt and is given by
pH=7 + 1/2(pKa-pKb)
Example 1. The pH of a 0.1-M solution of aniline, a weak base, is 8.8. Find its pKb.
Solution:
Given
The pH of a 0.1-M solution of aniline, a weak base, is 8.8.
⇒ \(\underset{\text { anline }}(\mathrm{B})+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{BH}^{+}+\mathrm{OH}^{-}\)
Initial conc.(M) C 0 0
Change -x +x +x
Equilibrium cone. C-x x x
Since \(K_{\mathrm{b}}=\frac{\left[\mathrm{BH}^{+}\right]\left[\mathrm{OH}^{-}\right]}{[\mathrm{B}]}\),
∴ \(K_b=\frac{x^2}{C-x}\).
The pH of the solution = – log[H3O+] = 8.8.
∴ [H3O+] = antilog (-8.8) =16 x 10-9 M.
Since we know that \(\left[\mathrm{OH}^{-}\right]=\frac{K_w}{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]},\)
⇒ \(\left[\mathrm{OH}^{-}\right]=\frac{1 \times 10^{-14}}{16 \times 10^{-9}}=6.25 \times 10^{-6}=x\)
Substituting the values of x and C in the expression for Kb we get
∴\(K_{\mathrm{b}}=\frac{\left(6.25 \times 10^{-6}\right)^2}{0.1-6.25 \times 10^{-6}}\)
Since 6.25 x 10-6 <<0.1, 0.1- 6.25 x 10-6 = 0.1
∴ \(K_{\mathrm{b}}=\frac{\left(6.25 \times 10^{-6}\right)^2}{0.1}=3.9 \times 10^{-10}\).
pKb =- log Kb =-log3.9 x 10-10
pKb =9.4
Example 2. The pKa of hypobromous acid (HOBr) is 8.7. Calculate the pH of a 0.1-M solution of the acid.
Solution:
Given
The pKa of hypobromous acid (HOBr) is 8.7.
⇒ \(\mathrm{HA}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{A}^{-}\)
Equilibrium cone. (M) C- x x x
Since \(K_4=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=\frac{x^2}{C-x}\),
pKa =-log Ka =8.7.
Ka =antilog(-8.7) = 2 x 10-9
Now substituting the value of Ka and C in the expression for Ka, we get
2 x 10-9 = \(\frac{x^2}{0.1-x}\)
As Ka is small, 0.1 >> x.
∴ 2 x 10-9 = \(\frac{x^2}{0.1}.\)
or x2 =2×10-9– x 0.1 = 2 x 10-10
“Le Chatelier’s principle MCQs with answers PDF”
or x \(=\sqrt{2 \times 10^{-10}}=1.4 \times 10^{-5} \mathrm{M} .\)
This is the concentration of H3O+ ions
[H3O+]=x=1.4 X 10-5 M.
pH =- log[H3O+] = 4.8.
The pH of a 0.1-M solution of HOBr is 4.8.
Example 3. Calculate the degree of ionisation of 0.1-M acetic acid in the presence of 0.1-M HCl The pKa of acetic acid is 4.74. Compare it with its ionisation in pure water.
Solution:
⇒ \(\mathrm{HAc}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{Ac}^{-}\)
⇒ \(K_{\mathrm{a}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{Ac}^{-}\right]}{[\mathrm{HAc}]}\)
or \(\mathrm{p} K_{\mathrm{a}}=-\log K_{\mathrm{a}}=4.74\)
or Ka = antilog(-4.74) =1.8 x 10-5.
Let us first calculate α in pure water.
If the equilibrium concentration of [H3O+] = Cα then that of Ac’ is also Cα and the concentration of acetic acid that remains undissociated is [HAc]= C(1 – α).
∴ \(K_4=\frac{(\mathrm{C} \alpha)(\mathrm{C} \alpha)}{C(1-\alpha)}\)
Since acetic add is a weak acid, α << 1
Hence \(K_{\mathrm{a}} \cong \frac{C^2 \alpha^2}{C}=C \alpha^2\)
or \(\alpha=\sqrt{\frac{K_a}{C}}=\sqrt{\frac{18 \times 10^{-5}}{0.1}}=0.013 .\)
Therefore the percentage dissociation is 1.3%. In the presence of 0.01-M HCl, the source of H3O+ is acetic acid as well as hydrochloric acid. The equilibrium concentrations are as shown below.
⇒ \(\begin{array}{ll}
\mathrm{HAc}+\mathrm{H}_2 \mathrm{O} & \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{Ac}^{-} \\
\mathrm{C}(1-\alpha) & \mathrm{C} \alpha+0.01 \quad \mathrm{C} \alpha
\end{array}\)
Now, \(K_{\mathrm{a}}=\frac{(C \alpha+0.01)(C \alpha)}{C(1-\alpha)}\)
We may neglect α in comparison to 1.
∴ \(\quad K_a \cong \frac{C^2 \alpha^2+0.01 C \alpha}{C} \)
or \(K_a=C \alpha^2+0.01 \alpha\)
or \(C \alpha^2+0.01 \alpha-K_a=0\).
This is a quadratic equation and a can be calculated using the formula
x = \(\frac{-b \pm \sqrt{b^2-4 a c}}{2 a}\)
∴\(\quad \alpha =\frac{-0.01 \pm \sqrt{(0.01)^2+4 \times C \times K_a}}{2 C}\).
Substituting for Ka and C, we get
α = \(\frac{-0.01 \pm \sqrt{(0.01)^2+4 \times 0.1 \times 18 \times 10^{-5}}}{2 \times 0.1}=\frac{-0.01 \pm 0.0104}{0.2} .\)
Discarding the negative root, a = 2 x 10-3 = 0.002
Percentage dissociation = 0.002 x 100 = 0.2%
This is far less than that in pure water. This shows that the equilibrium is affected in the presence of a common ion.
Buffer Solutions
Suppose you have a solution of a particular pH or a solution which has a certain concentration of H3O+ ions. If you add even a small amount of add or base to this solution, its pH will change—it will decrease if you add an add and increase if you add a base.
Both in nature and in industries, it is necessary to have solutions whose pH values do not change with the addition of a small amount of an add or a base.
The pH value of an ordinary solution may change even if it comes in contact with air. It can absorb carbon dioxide from the air and become more addic, for example.
- A solution which can resist change in pH when a small amount of an acid or a base is added to it (or when it is diluted) is called a buffer solution. The ability of such a solution to resist change in pH is called buffer action.
- A buffer solution can be acidic. Such a solution contains a weak acid and its salt with a strong base in equimolar quantities (a weak acid and a salt formed from its conjugate base), example, a solution of acetic acid and sodium acetate, which acts as a buffer solution around pH 4.75.
- A basic buffer solution, on the other hand, contains equimolar quantities of a weak base and its salt with a strong acid (a weak base and a salt formed from its conjugate base), example, NH4OH and NH4Cl, which acts as a buffer around pH 9.25.
- A buffer solution may also contain a single substance, generally the salt of a weak acid and a weak base, example, ammonium acetate (CH3COONH4).
- In order to understand how a buffer solution maintains a constant pH value, let us consider a solution containing equimolar quantities of CH3COOH (weak acid) and CH3COONa (a salt of acetic acid with a strong base).
In an aqueous solution acetic acid will be weakly ionised and sodium acetate will be mostly dissociated.
⇒ \(\mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})+\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})\)
⇒ \(\mathrm{CH}_3 \mathrm{COONa}(\mathrm{aq}) \longrightarrow \mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})+\mathrm{Na}^{+}(\mathrm{aq})\)
The solution will contain a large concentration of Na+ and CH3COO– ions, a very small concentration of H3O+ ions, and a large amount of undissociated acetic acid molecules.
If a few drops of an acid are added to this solution, the H3O+ ions provided by the acid disturb the equilibrium (between ionised and un-ionised acid), and CH3COO– ions in the solution combine with the H3O+ ions to counter the change.
⇒ \(\underset{\text { from buffer }}{\mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})}+\underset{\text { from acid }}{\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})} \rightleftharpoons \mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
Thus, the H3O+ ions provided by the acid are neutralised by CH3COO ions from the buffer solution and there is no change in pH.
If a few drops of a base are added to the solution, the OH” ions from the base combine with the H3O+ ions of the solution to form water.
⇒ \(\mathrm{OH}^{-}(\mathrm{aq})+\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq}) \longrightarrow 2 \mathrm{H}_2 \mathrm{O}(\mathrm{l})\)
- This disturbs the equilibrium (by reducing the concentration of H3O+ ions) and in accordance with Le Chatelier’s principle, more CH3COOH molecules ionise, restoring the original concentration of H3O+ ions or the pH.
- Blood is a natural buffer solution whose pH is about 7.4. The pH of sea water is also almost constant.
- Buffer solutions are required for electroplating and in the manufacture of photographic material and dyes, among many other industrial processes.
- The pH of culture media in biological laboratories also need to be resistant to change. The buffer solution of desired pH can be prepared by taking appropriate ratios of salt and acid or salt and base and by knowing the respective pKa and pKb values.
- An equation which is used to calculate the pH of a buffer solution is as follows.
pH = \(\mathrm{p} K_{\mathrm{a}}+\log \frac{[\text { salt }]}{\text { [acid] }}\) (for acidic buffer)
or pH = \(\mathrm{p} K_{\mathrm{a}}+\log \frac{[\text { base }]}{[\text { salt }]}\)
(for basic buffer)
This is called the Henderson-Hasselbalch equation. In the equation [acid], [base], and [salt] denote the concentrations of the acid, base, and salt respectively used to prepare the buffer solution.
Note that, in both the equations, pKa appears and it may be calculated for the conjugate acid of the base by using the expression pKa =pKw -pKb.
Example 1. Calculate the pH of an acetic acid-sodium acetate buffer in which the concentrations of the acid and salt are 0.5 M and 0.25 M respectively. Ka = 1.8x 10-5 for acetic acid.
Solution:
Using the Henderson-Hasselbalch equation,
pH = \(\mathrm{pK}_{\mathrm{a}}+\log \frac{\text { [salt] }}{\text { [acid] }}\)
pH = \(-\log \left(18 \times 10^{-5}\right)+\log \frac{0.25}{0.5}\) = 4.74 – 0.3010 = 4.44.
Example 2. Find the pH of an ammonia-ammonium chloride buffer solution in which the concentrations are \(C_{\mathrm{NH}_3}\)=0.2M and \(C_{\mathrm{NH}_4^{+}}\)= 0.3 M. Given that Ka for NH3 = 1. 76 x 10-5.
Solution:
pKa + pKb=pKw
or pKa=pKw-pKb
pKa = 14 – (- log 1.76 x 10-5) = 14-4.75 = 9.25.
Using this value of pKa in the equation
pH = \(\mathrm{p} K_{\mathrm{a}}+\log \frac{[\text { base] }}{[\text { salt }]}\)
pH = \(9.25+\log \frac{[0.2]}{[0.3]}\) = 9.25-0.18 = 9.07.
Example 3. Determine the pH of a buffer solution containing 0.03-M boric acid and 0.043-M sodium borate, given that pKa for B(OH)3 =9.00.
Solution:
pH = \(p K_{\mathrm{a}}+\log \frac{\text { [salt] }}{\text { [acid] }}\)
= \(9+\log \frac{0.04}{0.03}\) = 9 + 0.12 = 9.12.
Solubility Equilibria
The solubility of different salts in water or any other solvent varies to a large extent. For instance, calcium chloride is hygroscopic (absorbs water vapour from the atmosphere) in nature, and, on the other hand, lithium fluoride is almost insoluble.
There are certain factors which influence the solubility of salts in solvents. Common salt dissolves readily in water because the energies of coulmbic or ionic interaction between the two ions of a salt are overcome due to the high dielectric constant of water.
- Thus, in water Na+ and C- exist freely— the two do not attract each other. The dissolution of a salt in water has the effect of reducing the force between the ions, which then separate as a consequence.
- Ion solvation (solvation enthalpy—energy released in the process of solvation) also affects tire solubility of a salt in water or any other solvent. An ion is solvated when surrounded by several molecules of solvent.
- Solvation enthalpy is more for polar solvents and less for nonpolar solvents. Thus, salts do not dissolve in nonpolar solvents as the solvation enthalpy is not enough to overcome lattice enthalpy. Different salts exhibit different solubilities at different temperatures.
- Salts are divided into three categories based on solubility. Salts of solubility 0.1 M or greater are considered soluble.
- Salts of solubility between 0.1 M and 0.01 M are considered arc considered slightly soluble. And salts of solubility less than 0.01 M, though called insoluble, are actually sparingly soluble.
- A solid dissolves in a solvent until the solution and the solid are in equilibrium. The solution, at equilibrium, is saturated and its molar concentration is the molar solubility of the solid.
Solubility product: BaSO4 and AgCl arc sparingly soluble in water. When such a sparingly soluble salt is mixed with water, a small
amount of it dissolves and makes the solution saturated.
- The rest of it remains undissolved and an equilibrium is set up between the undissolved salt and the salt ions in solution.
- At equilibrium, the rate of dissolution of ions from the undissolved solid is equal to the rate of precipitation of ions from the saturated solution.
- You may have noticed that we are talking about an equilibrium between the undissolved solid and the ions in solution, and not the molecules in solution.
“Equilibrium constant (Kc and Kp) formula and examples”
This is because the small amount of the salt that dissolves gets completely dissociated into ions. The equilibrium can be represented as follows.
⇒ \(\underset{\text { or electrolyte }}Undissolved salt \rightleftharpoons ions in solution\)
Let us consider solid AgCl in contact with its saturated aqueous solution. The dissolution of the salt may be represented as
⇒ \(\mathrm{AgCl}(\mathrm{s}) \rightleftharpoons \mathrm{Ag}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
As in the other cases we have considered before, the equilibrium constant would be K= \(\frac{\left[\mathrm{Ag}^{+}\right]\left[\mathrm{Cl}^{-}\right]}{[\mathrm{AgCl}]}\)
The concentration of the undissolved salt is constant (it can be taken as 1) irrespective of the amount of the solid salt present. (This is true for any pure solid.)
Then K[AgCl] = [Ag+][C–] =Ksp
This constant (Ksp) is called the solubility product. In general, if a sparingly soluble salt Ax By dissociates in water, the equilibrium thus set up can be represented as
⇒ \(\mathrm{A}_x \mathrm{~B}_y \rightleftharpoons x \mathrm{~A}^{y+}+y \mathrm{~B}^{x-}\).
The solubility product may be expressed as Ksp = [Ay+]x[Bx-]y
The solubility product of a sparingly soluble salt (or electrolyte) at a given temperature may be defined as the product of the molar concentrations of its ions in a saturated solution, with each concentration term raised to the power equal to the number of ions of that species produced by the dissociation of one molecule of the electrolyte.
A few examples will make this clear.
⇒ \(\mathrm{Ca}_3\left(\mathrm{PO}_4\right)_2: K_{\text {sp }} =\left[\mathrm{Ca}^{2+}\right]^3\left[\mathrm{PO}_4^{3-}\right]^2\)
⇒ \(\mathrm{BaSO}_4: K_{\text {sp }} =\left[\mathrm{Ba}^{2+}\right]\left[\mathrm{SO}_4^{2-}\right]\)
⇒ \(\mathrm{Mg}\left(\mathrm{OH}_2\right): K_{\text {sp }}=\left[\mathrm{Mg}^{2+}\right]\left[\mathrm{OH}^{-}\right]^2\)
⇒ \(\mathrm{Ag}_2 \mathrm{CrO}_4: K_{\text {sp }} =\left[\mathrm{Ag}^{+}\right]^2\left[\mathrm{CrO}_4^{2-}\right]\)
- It is worthwhile to mention here that ionic product (or Qsp) is the product of the concentration of ions in any solution raised to their respective stoichiometries.
- It may have struck you that both solubility product and ionic products are products of the concentrations of ions in a solution (not considering the powers of the concentration terms for the moment).
- However, the ionic product has a more general application. It applies to both saturated and unsaturated solutions, while the solubility product applies only to saturated solutions.
- The ionic product is the same as the solubility product in a saturated solution.
- If the solubility product is greater than the ionic product, the solution is unsaturated.
- The higher the value of the solubility product the greater is the solubility of a salt.
- The ionic product cannot be greater than the solubility product. When it tends to exceed the value of the solubility product, precipitation (or combination of ions) starts.
If you know the solubility of a sparingly soluble salt at a particular temperature, you can easily calculate its solubility product.
Example 1. Find the solubility product of AgCl at T°C if its solubility at this temperature is 1.08 x 10-5 mol L-1.
Solution:
⇒ \(\mathrm{AgCl}(\mathrm{s}) \rightleftharpoons \mathrm{Ag}^{+}(\mathrm{aq})+\mathrm{Cl}^{-}(\mathrm{aq})\)
It is given that the solubility of AgCl is 1.08×10-5 mol L-1. AgCl is a sparingly soluble salt and it dissociates completely.
That is to say, 1 mol of AgCl in solution dissociates completely to produce 1 mol of Ag+ and 1 mol of Cl– ions.
Therefore, 1.08 x 10-5 mol of AgCl will give 108 x 10-5 mol of Ag+ and 1.08 x 10-5 mol of Cl– ions,
or [Ag+] = 1.08 x 10-5 mol L-1
and [Cl–] = 1.08x 10-5 mol L-1
= (1.08 x 10-5 )(1.08 x 10-5) = 1.16 x 10-10.
Example 2. The solubility of PbCl2 at a particular temperature is 2.2 x 10-2 mol L-1. Find its solubility product at this temperature.
Solution:
Given
The solubility of PbCl2 at a particular temperature is 2.2 x 10-2 mol L-1.
⇒ \(\mathrm{PbCl}_2 \rightleftharpoons \mathrm{Pb}^{2+}+2 \mathrm{Cl}^{-}\)
PbCl2 is a sparingly soluble salt, so it is completely ionised in solution.
In other words, 1 mol of PbCl2 in solution will dissociate to produce 1 mol of Pb+ and 2 mol of Cl– ions. It is given that the solubility of PbCl2 is 2.2 x 10-2 mol L-1.
[Pb2+] = 2.2 x 10-2 mol L-1
and [Cl–] = 2 x 2.2 x 10-2 mol L-1.
∴ Ksp =[Pb2+][Cl–]2 = [2.2 x 10-2][2 x 2.2 x 10-2]2 =4.3×10-5.
Calculation of solubility: The solubility of a salt is defined as grams of solute that can be dissolved in 100 mL of the solvent. The solubility of a sparingly soluble salt can be calculated if its solubility product is known.
- However, since Ksp is obtained considering the concentration in moles per litre (M), solubility is also obtained in moles per litre.
- This is called molar solubility—the amount of n salt that dissolves in a requisite amount of water to produce one litre of a saturated solution.
- The following examples illustrate the calculation of molar solubility from solubility product.
Example 1. The solubility product of Ag2CrO4 at 298 K is 4 x 10-12. Find its solubility at this temperature.
Solution:
Given
The solubility product of Ag2CrO4 at 298 K is 4 x 10-12.
At equilibrium \(\mathrm{Ag}_2 \mathrm{CrO}_4 \rightleftharpoons 2 \mathrm{Ag}^{+}+\mathrm{CrO}_4^{2-}\)
Suppose the solubility of Ag2CrO4 is x mol L-1
Then [Ag+]=2x and [CrO42-] = x. (Ag2CrO4 forms 2 moles of Ag+ and one mole of CrO42-)
∴ \(K_{\mathrm{sp}}=\left[\mathrm{Ag}^{+}\right]^2\left[\mathrm{CrO}_4^{2-}\right]=[2 x]^2[x]=4 x^3\)
Given that Ksp = 4 x 10-12
or 4x3 =4 x 10-12
∴ x = 1 x 10-4
Therefore the solubility of Ag2CrO4 is 1 x 10-4 mol L-1.
Example 2. The solubility product of AgBr in water is 2.5 x 10-13. Calculate its solubility in a 0.01-M NaBr solution.
Solution:
Given
The solubility product of AgBr in water is 2.5 x 10-13.
NaBr dissociates completely in solution.
∴ \(\left[\mathrm{Br}^{-}\right]=[\mathrm{NaBr}]_0=0.01 \mathrm{M}\) (The subscript ‘0’ denotes the initial concentration.)
Let the solubility of AgBr be x mol L-1
Then [Ag+] = [Br–] = x mol L-1 (from the dissociation of AgBr)
∴ total[Br] = x + 0.01 M (x <<0.01 M; AgBr being sparingly soluble)–
Ksp(AgBr) = [Ag+][B–r] = x x 0.01
Given that Ksp(AgBr) = 2.5 x 10-13
∴ x x 0.01 = 2.5 x 10-13
or x = \(\frac{2.5 \times 10^{-13}}{0.01}=2.5 \times 10^{-11} \mathrm{~mol} \mathrm{~L}^{-1}\)
Factors affecting solubility: The principle that an equilibrium counteracts the change due to the addition of species to solutions is also applicable to solubility product constants.
Common-ion effect: The presence of a common ion can also affect the solubility equilibrium. Let us consider the dissolution of MgF2 in water.
⇒ \(\mathrm{MgF}_2(\mathrm{~s}) \rightleftharpoons \mathrm{Mg}^{2+}(\mathrm{aq})+2 \mathrm{~F}^{-}(\mathrm{aq})\)
Let x be the solubility of MgF2 in moles per litre. Then the concentrations of Mg2+ and F– are as follows:
[Mg2+] = x mol L-1 (as one mole of MgF2 gives one mole of Mg2+ and 2 moles of F–).
[F–] = 2 xmol L-1.
∴ Ksp =[Mg2+][F–]2 = x x(2x)2 =4x3.
But the solubility product of MgF2 is 7.4 x 10-11
∴ 7.4 x 10-11 = 4x3
or \(x^3=\frac{7.4 \times 10^{-11}}{4}=1.8 \times 10^{-11} \Rightarrow x=\sqrt[3]{1.8 \times 10^{-11}}=2.6 \times 10^{-4} \mathrm{~mol} \mathrm{~L}^{-1}\)
∴ \({\left[\mathrm{Mg}^{2+}\right]=2.6 \times 10^{-4} \mathrm{~mol} \mathrm{~L}^{-1} .}\)
Molar solubility = 2.6 x 10-4 M.
∴ [F–] = 2 x 2.6 x 10-4 =5.2×10-4M.
When NaF is added to a solution of MgF2, F– being the common ion, its concentration in solution increases.
In order to keep Ksp constant, [Mg2+] must become smaller, or in other words, MgF2 is less soluble in an NaF solution than it is in pure water.
The presence of a common ion shifts the equilibrium to the left. This may be seen clearly from the following solved example.
Example: Calculate the molar solubility of MgF2 in 0.05-M NaF at 298 K, given that Kspof MgF2 is 7.4 x 10-11.
Solution:
We have calculated the molar solubility of MgF2 in pure water, which is 2.6×10-4 M. Let us now calculate its solubility in NaF.
If we assume x to be the molar solubility of MgF2 then one mole of MgF2 gives 1 mol of Mg2+ ions and 2 moles of F– ions.
Hence the concentrations of Mg2+ and F– from MgF2 alone are x and 2x respectively. There is another source of F–, which is NaF. From 0.05 M of NaF, we get 0.05 M of F– ions.
Equilibrium conc. \(\mathrm{MgF}_2(\mathrm{~s}) \rightleftharpoons \underset{x}{\mathrm{Mg}^{2+}(\mathrm{aq})}+\underset{2 x+0.05}{2 \mathrm{~F}^{-}(\mathrm{aq})}\)
Hence [F–] = 2x+ 0.05 and [Mg2+] = x.
But Ksp =[Mg2+][F–]2 or 7.4 x 10-11 = x x (2x + 0.05)2,
As Ksp is smqll 2x <<0.05.
Hence 7.4 x 10-11 s x x (0.05)2 or x = 7.4 x 10-9 M.
The molar solubility of MgF2 has decreased from 2.6 x 10-4 M in pure water to 7.4 x 10-9 M in the presence of NaF due to the common-ion effect.
pH of a solution: The solubility of salts of weak acids increases as the pH of the solution decreases or the acidity increases.
For example, the solubility of CaCO3 increases with decreasing pH.
This is because the CO32- ions react with H+ ions forming HCO3– ions react with H and thus are removed from the solution. As a result the reaction moves in the forward direction, dissolving more CaC03.
⇒ \(\begin{aligned}
\mathrm{CaCO}_3(\mathrm{~s}) \rightleftharpoons \mathrm{Ca}^{2+}(\mathrm{aq})+\mathrm{CO}_3^{2-}(\mathrm{aq}) \\
\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{CO}_3^{2-}(\mathrm{aq}) \rightleftharpoons \mathrm{HCO}_3^{-}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \\
\hline
\mathrm{CaCO}_3(\mathrm{~s})+\mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq}) \rightleftharpoons \mathrm{Ca}^{2+}(\mathrm{aq})+\mathrm{HCO}_3^{-}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \\
\hline
\end{aligned}\)
Salts containing anions such as \(\mathrm{PO}_4^{3-}, \mathrm{S}^{2-}, \mathrm{F}^{-} \text {and } \mathrm{CN}^{-}\) also behave in a similar fashion.
Predicting the precipitation of a salt: If the value of the ionic product exceeds that of the solubility product, precipitation of the excess ions occurs.
So, if you know the solubility product of a sparingly soluble salt, you will be able to predict whether or not mixing two solutions containing known concentrations of Us Ions will result in the precipitation of the salt.
Example 1. Predict whether precipitation will occur when equal volumes of a 0.02-M Na2SO4 solution and a 0.02-M BaC2 solution are mixed.(Ksp of BaSO4 = 1.5 x 10-10)
Solution:
Let the volume of each solution = V mL
Total volume after mixing = 2V mL
Applying the relation \(\begin{aligned}
M_1 V_1=M_2 V_2 \\
0.02 \times V=M_2 \times 2 V
\end{aligned}\)
∴ after mixing [BaCl2]0 = 0.01 mol L-1
Since BaCl2, is completely ionised,
[Ba2+] =[BaCl2]0 = 0.01 mol L-1
To determine the molarity of the Na2SO4 solution after mixing, we use the equation
M’1V’1(before mixing) = M’2V’2(after mixing)
∴ \(M_2^{\prime}=\frac{0.02 \times V}{2 V}=0.01\)
∴ after mixing[Na2SO4]0 = 0.01 mol L-1
Since Na2SO4is completely ionised,
[SO42-] = [Na2SO4] = 0.01 mol L-1
Ionic product of BaSO4, LP. or Qsp = [Ba2+][SO42-] = 0.01 x 0.01 = 10-4 mol L-1
As Qsp > Ksp precipitation occurs.
Precipitation of soluble salts: The precipitation of soluble salts from their saturated solutions is called salting out This process is used to obtain pure sodium chloride from a saturated solution of impure sodium chloride.
- HCl gas is passed through a saturated solution of NaCL The addition of HCl (which ionises) increases the concentration of Cl– ions.
- Consequently, the ink product exceeds the solubility product of NaCl and precipitation occurs. The salt which is precipitated is in the pure stale. The impurities remain in solution.
- Soap is salted out from its saturated solution by the addition of sodium chloride.
- Soaps are sodium salts of ratty acids. The addition of sodium chloride increases the concentration of Na+ ions in the solution, thus making the ionic product exceed the solubility product
Example 1. Calculate the solubility of BaSO4 in pure water and in sea -water which contains 0.029-M SO42- ions. [Ksp(BaSO4) = 9.1x 10-11n. ]
Solution:
⇒ \(\mathrm{BaSO}_4(\mathrm{~s}) \rightleftharpoons \mathrm{Ba}^{2+}(\mathrm{aq})+\mathrm{SO}_4^{2-}(\mathrm{aq})\)
Let x be the molar solubility of BaSO4.
∴ \(\left[\mathrm{Ba}^{2-}\right]=x ;\left[\mathrm{SO}_4^{2-}\right]=x\)
⇒ [latexK_{\mathrm{sp}}=\left[\mathrm{Ba}^{2+}\right]\left[\mathrm{SO}_4^{2-}\right]=x^2 .[/latex]
But \(9.1 \times 10^{-11}=x^2 \text { or } x=\sqrt{9.1 \times 10^{-11}}=9.5 \times 10^{-6}\) .
Hence the solubility of BaSO4 in pure water = 9.5 x 10-6 M
In sea water, [SO42-] = 0.029 M.
Total concentration of SO42- = 0.029 + x’, where x’ is the solubility of BaSO4 in sea water.
Now, Ksp = (x’)(0.029 + x’)
We may assume that x'<< 0.029 as Ksp is small.
∴ Ksp =(x’)(0.029)
∴or 9.1 x 10-11 = 0.29x’
or x’ = 3.1x 10-10
Therefore, the solubility of BaSO4 in sea water is 3.1 x 10-10 M. This is far less than that in pure water.
Example 2. A carbonated drink was saturated with CO2 at 0°C and 5 atm pressure. What was the concentration of CO2 in the bottled drink if the Henry constant for an aqueous solution of CO2 at 0°C is 7.5 x 10-2 mol L-1 atm-1?
Solution:
Given
A carbonated drink was saturated with CO2 at 0°C and 5 atm pressure.
According to Henry’s law, C = kp,
where C is the concentration of a gas in a solution, p is the partial pressure of the gas and k is the Henry constant.
∴ concentration of CO2 in the drink = kp = 7.5 x 10-2 x5
=37.5 x 10-2 =0.38 mol L-1.
Example 3. Calculate the equilibrium constantfor a reaction given that the rate constantsfor thefonvard and reverse reactions are 2.48 x 10-4 and 8.25 x10-5 respectively.
Solution:
Equilibrium constant K = \(\frac{k}{k^{\prime}}\),
where k and k’ are the rate constants for the forward and the reverse reaction respectively.
∴ K = \(=\frac{2.48 \times 10^{-4}}{8.25 \times 10^{-5}}=3\)
Example 4. Two moles of PCl5 was heated to 327°C in a closed vessel of volume 2 L. When equilibrium was established, 40% of the PCl5 had dissociated into PCI3 and Cl2 Calculate the equilibrium constantfor the reaction.
Solution:
Given
Two moles of PCl5 was heated to 327°C in a closed vessel of volume 2 L. When equilibrium was established, 40% of the PCl5 had dissociated into PCI3 and Cl2
The dissociation of PCl5 into PCI3 and Cl2 can be represented as follows.
⇒ \(\mathrm{PCl}_5 \rightleftharpoons \mathrm{PCl}_3+\mathrm{Cl}_2\)
Initial concentration of PCl5 = 2 mol.
Percentage dissociation at equilibrium = 40%.
PCI5 dissociated at equilibrium = 40% of 2 mol = 0.8 mol.
∴ amount of PCI5 at equilibrium = 2- 0.8 =12 mol
and amount of PCI3 and Cl2 at equilibrium = 0.8 mol.
Since the volume of the vessel is 2 L, at equilibrium
⇒ \(\left[\mathrm{PCl}_5\right]=\frac{1.2}{2} \mathrm{~mol} \mathrm{~L}^{-1}\)
⇒ \(\left[\mathrm{PCl}_3\right]=\frac{0.8}{2} \mathrm{~mol} \mathrm{~L}^{-1}\).
⇒ \(\left[\mathrm{Cl}_2\right]=\frac{0.8}{2} \mathrm{~mol} \mathrm{~L}^{-1}\).
∴ K=\(\frac{\left[\mathrm{PCl}_3\right]\left[\mathrm{Cl}_2\right]}{\left[\mathrm{PCl}_5\right]}=\frac{0.4 \times 0.4}{0.6}=0.267\).
Example 5. 0.1 mol of PCl5 is heated in a 1-L flask at 250°C. Calculate the equilibrium concentrations of PCl5, PCl3 and Cl2, if the equilibrium constant for the dissociation of PCl5 is 0.0414.
Solution:
Given
0.1 mol of PCl5 is heated in a 1-L flask at 250°C. Calculate the equilibrium concentrations of PCl5, PCl3 and Cl2,
PCI5 dissociates according to the following reaction.
∴ \(\mathrm{PCl}_5 \rightleftharpoons \mathrm{PCl}_3+\mathrm{Cl}_2\)
Suppose at equilibrium x mol of PCl5 dissociates to form x mol of PCl3 and x mol of Cl2. Since the volume of the vessel is 1 L, at equilibrium,
⇒ [PCl5] = 0.1 – x mol L-1.
⇒ [PCl3] = xmolL
⇒ [Cl2] = x mol L-1.
∴ K = \(\frac{\left[\mathrm{PCl}_3\right]\left[\mathrm{Cl}_2\right]}{\left[\mathrm{PCl}_5\right]}=\frac{x \times x}{0.1-x}\)
Given that K = 0.0414.
∴ 0.0414 = \(\frac{x^2}{0.1-x}\)
or x2 +0.0414x-0.00414 = 0
This is a quadratic equation and x can be calculated using the formula
x =\(-\frac{b \pm \sqrt{b^2-4 a c}}{2 a}=\frac{-0.0414 \pm \sqrt{(0.0414)^2-4 \times 1 \times(-0.0414)}}{2 \times 1}\)
=\(\frac{-0.0414 \pm \sqrt{0.0017+0.1656}}{2}=\frac{-0.0414 \pm 0.135}{2}\) = 0.0882 and + 0.0468
The negative value of x is meaningless.
Thus, the concentrations of PCI3, Cl2 and PCl5 at equilibrium are as follows.
[PCI3] = [Cl2] = x = 0.0468 mol LT-1.
[PCI5] = 0.1 – x = 0.0532 mol L-1.
Example 6. The equilibrium constant for the reaction \(\mathrm{CH}_3 \mathrm{COOH}+\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH} \rightleftharpoons \mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5+\mathrm{H}_2 \mathrm{O}\) is 4.0 at 298 K. Calculate the weight of ethyl acetate that will be obtained when 120 g of acetic acid reacts with 92 g of alcohol.
Solution:
Given
The equilibrium constant for the reaction \(\mathrm{CH}_3 \mathrm{COOH}+\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH} \rightleftharpoons \mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5+\mathrm{H}_2 \mathrm{O}\) is 4.0 at 298 K.
Weight of ethyl alcohol = 92 g.
Weight of acetic acid = 120 g.
Number of moles = \(\frac{\text { weight of substance }}{\text { molecular weight }} \text {. }\)
∴ number of moles of ethyl alcohol = 92/46 = 2
and number of moles of acetic acid = 120/60 = 2.
According to the chemical equation that represents the reaction, 1 mol each of the reactants produce 1 mol each of the products. Therefore, if x mol of each of the reactants reacted, x mol each of the products would be formed at equilibrium.
⇒ \(\mathrm{CH}_3 \mathrm{COOH}+\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH} \rightleftharpoons \mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5+\mathrm{H}_2 \mathrm{O}\)
“Effect of temperature and pressure on chemical equilibrium”
Intial concentration \(\frac{2}{V} \frac{2}{V} \frac{0}{V} \frac{U}{V}\)
Conc. at equilibrium \(\frac{2-x}{V} \frac{2-x}{V} \frac{x}{V} \frac{x}{V}\)
Applying the law of equilibrium
K = \(\frac{\left[\mathrm{CH}_3 \mathrm{COOC}_2 \mathrm{H}_5\right]\left[\mathrm{H}_2 \mathrm{O}\right]}{\left[\mathrm{CH}_3 \mathrm{COOH}\right]\left[\mathrm{C}_2 \mathrm{H}_5 \mathrm{OH}\right]}\)
= \(\frac{[x / V][x / V]}{\left[\frac{2-x}{V}\right]\left[\frac{2-x}{V}\right]}=\frac{x^2}{(2-x)^2}\).
Given that K = 4.
∴ 4 = \(\frac{x^2}{(2-x)^2}\)
or 2 = \(\frac{x}{2-x}\)
or 4-2x = x
or x = 4/3 =1.33 mol.
Hence the amoimt of ethyl acetate formed = 1.33 mol
=1.33 x molecular weight of ethyl acetate
=1.33 x 88 =117.04 g
Example 7. The equilibrium constant at 298 K for \(\mathrm{Cu}(\mathrm{s})+2 \mathrm{Ag}^{+}(\mathrm{aq}) \rightleftharpoons \mathrm{Cu}^{2+}(\mathrm{aq})+2 \mathrm{Ag}(\mathrm{s})\) is 4.0 x 1015. In a solution in which copper has displaced some silver ions, the concentration of Cu2+ ions is 16 x 10-2 mol L-1 and the concentration of Ag+ ions is 2.0 x 10-9 mol L-1. Is this system at equilibrium?
Solution:
Given
The equilibrium constant at 298 K for \(\mathrm{Cu}(\mathrm{s})+2 \mathrm{Ag}^{+}(\mathrm{aq}) \rightleftharpoons \mathrm{Cu}^{2+}(\mathrm{aq})+2 \mathrm{Ag}(\mathrm{s})\) is 4.0 x 1015. In a solution in which copper has displaced some silver ions, the concentration of Cu2+ ions is 16 x 10-2 mol L-1 and the concentration of Ag+ ions is 2.0 x 10-9 mol L-1.
Applying the law of chemical equilibrium, assuming that the system is at equilibrium,
⇒ \(K_{\mathrm{c}}=\frac{\left[\mathrm{Cu}^{2+}(\mathrm{aq})\right][\mathrm{Ag}(\mathrm{s})]^2}{\left[\mathrm{Ag}^{+}(\mathrm{aq})\right]^2[\mathrm{Cu}(\mathrm{s})]} .\)
By convention, concentration of a solid = 1.
∴\(K_{\mathrm{c}}=\frac{\left[\mathrm{Cu}^{2+}(\mathrm{aq})\right]}{\left[\mathrm{Ag}^{+}(\mathrm{aq})\right]^2}\)
Substituting the concentrations of Cu2+ and Ag+ ions
∴ \(K_c=\frac{1.6 \times 10^{-2}}{\left(2 \times 10^{-9}\right)^2}=4 \times 10^{15}\)
This is the value of Kc for the reaction in equilibrium. Hence the given system is in equilibrium.
Example 8. The equilibrium constant for the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\) is 50.5 at 718 K. Predict the direction in which the reaction will proceed to reach equilibrium at 718 K if we start with 2 x 10-2 mol of HI,1.0 x 10-2 mol of H2 and 3.0 x 10-2 mol of I2 in a vessel of volume1 L
Solution:
Given
The equilibrium constant for the reaction \(\mathrm{H}_2(\mathrm{~g})+\mathrm{I}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{HI}(\mathrm{g})\) is 50.5 at 718 K.
Reaction quotient \(\left(\mathrm{Q}_c\right)=\frac{[\mathrm{HI}]^2}{\left[\mathrm{H}_2\right]\left[\mathrm{I}_2\right]}\)
If the reaction quotient is equal to Kc, the system is at equilibrium, and if it is less than Kc, the reaction will proceed in the forward direction to attain equilibrium.
Since the volume of the vessel is 1 L,
⇒ \(Q_c=\frac{\left[2 \times 10^{-2}\right]^2}{\left[1.0 \times 10^{-2}\right]\left[3.0 \times 10^{-2}\right]}=1.33\)
Since this is less than Kc, the reaction will proceed in the forward direction.
Example 9. 1 mol of ammonia was injected into a 1-L flask at a certain temperature. The equilibrium mixture was found to contain 0.3 mol of H2. Calculate the concentrations of N2 and NH3 at equilibrium, and the equilibrium constant.
Solution:
Given
1 mol of ammonia was injected into a 1-L flask at a certain temperature. The equilibrium mixture was found to contain 0.3 mol of H2. Calculate the concentrations of N2 and NH3 at equilibrium
The equilibrium can be represented as follows.
⇒ \(2 \mathrm{NH}_3 \rightleftharpoons \mathrm{N}_2+3 \mathrm{H}_2\)
The initial concentration of NH3 =1 mol.
At equilibrium, 2 mol of ammonia produces 3 mol of hydrogen.
Therefore, 0.3 mol of H2 will be obtained from 0.2 mol of NH3.
Similarly, when 2 mol of NH3 dissociates, 1 mol of N2 is obtained.
Therefore, 0.2 mol of NH3 will produce 0.1 mol of N2.
Since the volume of the flask is1 L, at equilibrium,
[N2] = 0.1 mol -1,
[H2] = 0.3 mol L-1,
[NH3] =1-0.2 =0.8 mol L-1.
∴ \(K_{\mathrm{c}}=\frac{\left[\mathrm{N}_2\right]\left[\mathrm{H}_2\right]^3}{\left[\mathrm{NH}_3\right]^2}=\frac{(0.1)(0.3)^3}{(0.8)^2}=0.004\)
Example 10. 1 mol of ammonium carbamate dissociates as shown below at 500 K. \(\mathrm{NH}_2 \cdot \mathrm{COONH}_4(\mathrm{~s}) \rightleftharpoons 2 \mathrm{NH}_3+\mathrm{CO}_2\) If the pressure exerted by the released gases is 3.0 bar, calculate the value of Kp.
Solution:
Applying the law of equilibrium,
⇒ \(K_p=\frac{p_{\mathrm{NH}_3}^2 p_{\mathrm{CO}_2}}{p_{\mathrm{NH}_2 \cdot \mathrm{COONH}_4(s)}}\)
By convention \(p_{\mathrm{NH}_2 \mathrm{COONH}}\) =1 as it is a solid.
Given that the pressure exerted by the gases = 3 bar; this is the total pressure.
(To obtain the partial pressures, the total pressure must be multiplied by the mole fractions of the respective gases.)
∴ \(p_{\mathrm{NH}_3}=3 \times \frac{2}{3}=2 \mathrm{bar}\).
and \(p_{\mathrm{CO}_3}=3 \times \frac{1}{3}=1 \mathrm{bar}\).
∴ \(K_p=(2)^2(1)=4 bar.\)
Example 11. The dissociation constant of a weak acid HA is 1.6 x 10-5 at 298 K. Calculate the H3O+ ion concentration in a 0.01-M solution of the acid.
Solution:
Given
The dissociation constant of a weak acid HA is 1.6 x 10-5 at 298 K.
The ionisation of the acid may be represented as
⇒ \(\mathrm{HA}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l}) \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}(\mathrm{aq})+\mathrm{A}^{-}(\mathrm{aq})\)
⇒ \(K_a=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\) .
If the concentration of HA that dissociates is x, then the concentration of H3O+ and A– is also x.
Then the concentration of the undissociated acid is (0.01- x).
Since HA is a weak acid, its degree of dissociation is small and at equilibrium, x may be neglected in comparison to 0.1.
[HA] = (0.1 -x) = 0.1 mol L-1
[H3O+] =[A–] = X
∴ \(\quad K_a=\frac{x^2}{0.1}\)
or \(x^2=K_a \times 0.1=16 \times 10^{-5} \times 0.1\)
or \(x=\sqrt{16 \times 10^{-5} \times 0.1}=126 \times 10^{-3}\).
∴ \(\quad\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\left[\mathrm{A}^{-}\right]=1.26 \times 10^{-3} \mathrm{~mol} \mathrm{~L}^{-1}\).
Example 12. Calculate the degree of dissociation and the concentration of H3O+ ions in a 0.1-M solution of formic acid, given that =2.1 x 10-4 at 298 K.
Solution:
Formic acid dissociates in water according to the following equation.
⇒ \(\mathrm{HCOOH}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{HCOO}^{-}\)
The initial concentration of the acid is given as 0.1 mol L-1
Let the degree of dissociation be α.
Then at equilibrium
[H3O+] = α x 0.1 mol L-1
[HCOO–] =α x 0.1 mol L-1
[HCOOH] = 0.1(1- α) mol -1
∴\(K_a=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{HCOO}^{-}\right]}{[\mathrm{HCOOH}]}=\frac{(0.1 \alpha)(0.1 \alpha)}{0.1(1-\alpha)}\)
Since formic add is a weak acid, a is very small as compared to 1 and can be neglected in the denominator.
∴ \(K_a=\frac{(0.01 \alpha)^2}{0.1}=0.1 \times \alpha^2\)
Given that Ka = 2.1 x 10-4.
or 2.1 x 10 = 0.1 x α2
or \(\frac{21 \times 10^{-4}}{0.1}=\alpha^2\)
or α2 = 2.1 x 10-3 = 21 x 10-4
or α = 0.046
[H3O+] = Cα = 0.1 x 0.046 = 4.6 x 10-3 mol L-1
Example 13. A solution of NaOH contains 4.0 g of the base per litre. Find the pH of the solution.
Solution:
Concentration of NaOH solution = 4 g L-1; molecular weight of NaOH = 40.
∴ molarity of solution = 4/40 = 0.1
Since NaOH is completely dissociated, concentration of OH– ions = 0.1 M = 10-1 mol L-1.
Kw=[H3O+][OH–]
or 1 x 10-14 = [H3O+][OH–]
or \(\frac{1 \times 10^{-14}}{10^{-1}}=\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\)
or 1 x 10-13 M = [H3O+].
pH =- log[H3O+] = -log 10-13 = 13.
Example 14. Calculate the pH of a solution of a strong monobasic acid made by diluting 100 mL of a 0.01-M solution to 1 L
Solution:
M1V1 = M2V2
or 0.01 x100 = M2 x 1000.
∴ \(M_2=\frac{0.01 \times 100}{1000}=10^{-3}\)
Assuming complete dissociation
[H3O+] =10-3. (it is a monobasic add, one mole of protons is released per mole of add.)
∴ pH =- log[H3O+] =-[log10-3] = 3.
Example 15. Calculate the pH of a
- 0.02-M H2SO4 solution and
- 0.01-M Ba(OH)2 solution.
Solution:
1. The ionisation of H2SO4 can be represented by \(\mathrm{H}_2 \mathrm{SO}_4+2 \mathrm{H}_2 \mathrm{O} \longrightarrow 2 \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{SO}_4^{2-}\)
Being a strong acid, H2SO4 is completely ionised.
1 mol of H2SO4 dissodates to give 2 mol of H3O+.
[H3O+] =2 x 0.02 = 0.04 mol L-1.
∴ pH = -log 0.04 =-log 4 x 10-2 = -(-1.3980) =1.398.
2. Ba(OH)2 dissodates completely.
1 mol of Ba(OH)2 dissociates to give 2 mol of OH–.
∴ [OH–] = 2[Ba(OH)2] =2 x 0.01 = 0.02 M.
Kw =[OH–][H3O+]
∴ \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\frac{\mathrm{K}_{\mathrm{w}}}{\left[\mathrm{OH}^{-}\right]}=\frac{1 \times 10^{-14}}{0.02}=5 \times 10^{-13} \mathrm{M}\)
∴ pH =-log 5 x 10-13 =-(-12. 3010) =12.301
Example 16. How many grams of NaOH must be dissolved in 1 L of a solution of the base so that the pH of the solution is 13?
Solution:
pH = -log[H3O+]
[H3O+] = -antilog pH = -antilog13 =1 x 10-13 mol L-1.
Kw =[H3O+][OH–].
∴ \(\left[\mathrm{OH}^{-}\right]=\frac{K_{\mathrm{w}}}{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}=\frac{1 \times 10^{-14}}{1 \times 10^{-13}}=1 \times 10^{-1} \mathrm{~mol} \mathrm{~L}^{-1}\).
As NaOH is a strong electrolyte, [NaOH] =[OH–] =1 x 10-1 mol L-1.
Amount of NaOH = number of moles x mol. wt. of NaOH =1 x10-1 x 40 = 4 g.
Example 17. The dissociation constant of a weak acid HA is 1 x 10-9. Find the pH of a 0.1-M solution of the acid.
Solution:
The dissociation of the acid in water may be represented as follows.
⇒ \(\mathrm{HA}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{A}^{-}\)
If x mol dissociates at equilibrium, [HA] = 0.1- x.
∴ \({\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\left[\mathrm{A}^{-1}\right]=x .}\)
∴ \(\mathrm{K}_{\mathrm{a}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=\frac{x \times x}{0.1-x}\).
Since x is negligible compared to 1,
∴ \(K_{\mathrm{a}}=\frac{x^2}{0.1}\)
∴ x = \(\sqrt{\mathrm{K}_{\mathrm{a}} \times 0.1}=\sqrt{1 \times 10^{-9} \times 0.1}=10^{-5} \mathrm{~mol} \mathrm{~L}^{-1}\)
∴ \({\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=10^{-5} \mathrm{~mol} \mathrm{~L}^{-1} .}\)
∴ \(\mathrm{pH}=-\log \left[\mathrm{H}_3 \mathrm{O}^{+}\right]=-\log 10^{-5}=5\) .
Example 18. Calculate, the[H3O+] of a solution of a monobasic acid whose pH is 4.35.
Solution:
pH = -log[H3O+]
∴ log[H3O+] = -pH = -4.35.
[H3O+ ] = antilog(-4.35).
Since mantissas are always positive in logarithmic tables, add -1 to the characteristic and +1 to mantissa in order to make the mantissa positive.
[H3O+] = antilog[-4-1 + (-0.35 +1)] = antilog 5.65 = 4.467 x 10-5
Equilibrium Multiple Choice Questions
Question 1. Which of the following is a characteristic of a reversible reaction?
- The number of moles of the reactants is equal to that of the products.
- It can be influenced by a catalyst.
- It can never proceed to completion.
- None of the above
Answer: 3. It can never proceed to completion.
Question 2. A chemical reaction A ⇔ B is said to be in equilibrium when
- A has been completely converted to b
- The conversion of a to b is 50%
- Only 10% of the conversion of a to b has taken place
- The rate of conversion of a to b is just equal to that of b to a
Answer: 4. The rate of conversion of a to b is just equal to that of b to a
Question 3. The equilibrium constants of these reactions are related as
⇒ latex]2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) \rightleftharpoons 2 \mathrm{SO}_3(\mathrm{~g}) K_1[/latex]
⇒ \(2 \mathrm{SO}_3(\mathrm{~g}) \rightleftharpoons 2 \mathrm{SO}_2(\mathrm{~g})+\mathrm{O}_2(\mathrm{~g}) K_2\)
- K2 = K1
- \(K_2=K_1^2\)
- \(K_2=\frac{1}{K_1^2}\)
- \(K_2=\frac{1}{K_1}\)
Answer: 4. \(K_2=\frac{1}{K_1}\)
Question 4. In which of the following cases does the reaction proceed the fastest?
- K =102
- K = 10-2
- K = 10
- K = 1
Answer: 1. K =102
Question 5. For the reaction \(\mathrm{PCl}_5(\mathrm{~g}) \rightleftharpoons \mathrm{PCl}_3(\mathrm{~g})+\mathrm{Cl}_2(\mathrm{~g})\), the forward reaction at constant temperature is favoured by
- The introduction of an inert gas at constant volume
- The introduction of an inert gas at constant pressure
- The introduction of Cl2 at constant volume
- An increase in pressure
Answer: 2. The introduction of an inert gas at constant pressure
Question 6. In the reaction \(2 \mathrm{~A}(\mathrm{~g})+\mathrm{B}(\mathrm{g}) \rightleftharpoons \mathrm{C}(\mathrm{g})+85 \mathrm{cal}\), which of the following conditions would give the best yield at equilibrium?
- 500 atm, 100°C
- 100 atm, 100°C
- 500 atm, 500°C
- 100 atm, 500°C
Answer: 1. 500 atm, 100°C
Question 7. Tire solubility of Agl in a solution of Nal is less than that in pure water because
- Agl forms a complex with nal
- Of the common-ion effect
- The Ksp of AgI is less than that of nal
- Of a decrease in temperature
Answer: 2. Of the common-ion effect
Question 8. Which of the following is not a Lewis acid?
- BF3
- AlCl3
- BeCl2
- SnCl4
Answer: 4. SnCl4
Question 9. An acid solution of pH 6 is diluted a hundred times. The pH of the solution becomes
- 6.95
- 6
- 4
- 9
Answer: 1. 6.95
Question 10. If the solubility of a salt M2X3 is x mol L-1, its solubility product is
- x5
- 76x2
- 96x5
- 108x2
Answer: 4. 108x2
Question 11. The solubility product of A2B is 4 x 10-9 mol L-1 Its solubility is
- 10-3 M
- 41/3 x 10-3 M
- 10-4 M
- 2 x 10-5 M
Answer: 1. 10-3 M
Question 12. Which of the following has the lowest value of Ksp at ordinary temperature?
- Mg(OH)2
- Ba(OH)2
- Ca(OH)2
- Be(OH)2
Answer: 4. Be(OH)2
Question 13. Which of the following is the strongest Bronsted base?
- ClO–
- ClO2–
- ClO3–
- ClO4–
Answer: 1. ClO–
Question 14. The pH of a certain solution is 5.0. Just enough acid is added to decrease the pH to 2.0. The hydrogen ion concentration will
- Increase 1000 times
- Decrease 1000 times
- Increase 100 times
- Increase 10 times
Answer: 1. Increase 1000 times
Question 15. The Ksp of CaF2 is 1.7 x 10-10. The addition of what concentration of an equal volume of F– ions to a solution containing 0.01-M Ca2+ ions will result in the precipitation of CaF2?
- 3.4×10-8 M
- 1.3×10-4 M
- 3.68 x 10-4 M
- 2.6×10-4 M
Answer: 2. 1.3×10-4 M